
Antioxidative activity of methyl amygdalinate from the seeds of Prunus persica and neuroprotective effects on Aβ1–42-induced neurodegeneration models†
Abstract
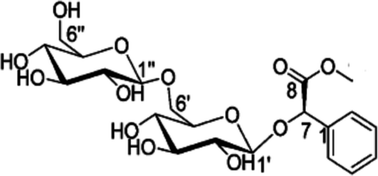
Prunus persica has been frequently used as a functional and medicinal food in China and other Asian countries. Aromatic glucosides isolated from the dried seeds of P. persica have been reported to possess antioxidant activity. The aim of this study was to investigate methyl amygdalinate (MAM), an active aromatic glucoside from P. persica with antioxidative and neuroprotective activities, in an Aβ1–42-induced mouse model of Alzheimer's disease. Neuroprotective effects of MAM (10, 100 μg kg−1) were estimated through the spontaneous locomotor test and the Morris water maze test. Antioxidative effects of MAM were evaluated by measurement of SOD and MDA levels in the hippocampus and cerebral cortex of model mice. In addition, the expressions of BDNF (neurotrophic factor), JKN/p38 (both contribute to MAPKs signaling pathways) in mouse brain were measured. Histopathological examination was used to represent the potential mechanisms. It was found that intracerebroventricular (i.c.v.) administration of MAM to Aβ1–42-induced mice significantly increased the swimming time in the target quadrant in the Morris water maze test. It also noticeably decreased SOD activity, and restored MDA level both in the hippocampus and cerebral cortex in mice. Furthermore, MAM could change the expressions of BDNF, and inhibit the expression of JKN/p38 in mouse brain. Results of histopathological examination also indicated that MAM markedly ameliorated neurodegeneration in the hippocampus in mice. In summary, MAM might protect against cognitive deficits and neurodegeneration by releasing the damage of oxidative stress and inhibiting the MAPKs inflammatory signaling pathways.
 Please wait while we load your content...
Please wait while we load your content...

