
Latest trends in molecular imprinted polymer based drug delivery systems
Shabi Abbas Zaidi
Department of Chemistry, Kwangwoon University, Wolgye-Dong, Nowon-Gu, Seoul, 139-701, Korea. E-mail: shabizaidi79@gmail.com; Fax: +82-2-911-8584; Tel: +82-2-940-8661
First published on 5th September 2016
Abstract
Molecular imprinted polymers (MIP) are promising and versatile materials that have been used for the determination of many different analytes. In the last few years, MIPs have been substantially employed for various biomedical applications, especially drug delivery systems (DDS), owing to some of their unique features such as specific recognition by imprinting the desired analyte, suitability in rough experimental conditions, and targeted and sustained drug release. Hence, this review is focused on the development of strategies undertaken for their application to drug delivery systems involving several different administration routes (i.e. transdermal, ocular and oral routes) published between 2014 and now. Herein, we have also highlighted the summaries of published works, in order to gain a better understanding of the synthetic strategies employed and the analytical performances of the reported MIPs, in addition to pointing out the challenges and future perspectives of MIP based DDS.
Shabi Abbas Zaidi has been an Assistant Professor in the Department of Chemistry, Kwangwoon University, Korea, since 2012. He obtained his B.Sc. and M.Sc. from Aligarh Muslim University, Aligarh, India and Ph.D. in Analytical Chemistry from Inha University, Korea, in 2009. His research interests cover molecular imprinting technologies, synthesis of nanomaterials and composite materials and their application to electrochemical sensors, biosensors and drug delivery. |
Introduction
Molecular imprinted techniques (MITs) are considered useful in obtaining highly selective polymeric materials called molecular imprinted polymers (MIPs). In the 1930s, it was the innovative work of Polyakov using silica matrices, that demonstrated the affinity of polymers for templates during polymerization.1 For a few decades, this phenomenon did not attract due attention and became dormant. The term “MIP” was coined in the 70s, when the pioneering research of Wulff & Sarhan2 and Arshady & Mosbach3 brought this field back into focus. With the continuous development in synthetic methods, characterization analysis and the utilization of MIPs in various fields over recent years, several excellent ways to imprint a wide range of target molecules have been developed. Consequently, the scientific community has shown a tremendous interest in exploiting the unique features of MIPs, including high selectivity, high stability and good durability against harsh conditions (e.g. thermal, mechanical, and highly acidic and basic pH conditions) for various purposes.In the general preparation protocol of an MIP, a cocktail solution containing a template (target) molecule, a monomer, and a cross-linker are dissolved in an appropriate solvent, resulting in a highly cross-linked polymer due to new bond formation between the template and cross-linked polymer during the curing process. Later, the imprinted template molecules are extracted from the as-synthesized polymer using a solvent mixture of a simple organic solvent and a mild acid or Soxhlet apparatus. Permanent nano-sized cavities of the original template are then generated in the polymer matrix corresponding to the shape, size and orientation of the template molecules. Due to the selective conformations of the shape, size, corresponding bond formations and surface chemistry, these cavities are capable of fitting template molecules preferentially over structural analogues.4 The working mechanism of MIPs is similar to the concept of the “lock and key” hypothesis of protein–guest interaction, which is a very well-known principle in the understanding of antibody–antigen interactions.5–7
There are two possible ways to prepare MIPs: covalent and non-covalent procedures, where either the covalent or non-covalent characteristics of the interaction between the template and the cavity are favored, respectively. It is obvious that the removal of the template from a covalently bonded polymer and rebinding the template to the cavity are not easy tasks, unless the MIP is obtained via non-covalent bonds, as these are weaker. In the general preparation method of an MIP, five different components are required: the template, the functional monomer, the cross-linker, the porogen and the initiator. The template and its functionalities usually determine the choice of the functional monomer. The polymer is prepared via a standard free radical polymerization initiated either by thermal or UV light, or electropolymerization might be used, depending on the reaction conditions. The amount of prepolymer constituents, polymer reaction times, temperature and the amount of porogen (i.e. organic solvents or water in aqueous preparations) are responsible for determining and regulating the morphology of the polymer. Finally, the as-fabricated imprinted cavities are capable of rebinding the imprinted molecules selectively.8,9
Owing to some of the remarkable properties of MIPs, considerable research endeavors have been dedicated to the advancement of MIPs, and during this time MIPs have shown great potential in diverse fields. MIPs have been implemented in numerous applications such as the recognition of biomolecules,10–12 protein detection,13,14 proteomic analysis,15 chromatographic separation,16–21 capture of hazardous radioactive waste,22 solid phase extraction,23,24 recognition of elements in electrochemical sensors and biosensors,25,26 and drug delivery.27–29 Stimuli-responsive (i.e. pH and temperature) MIPs have been extensively utilized in various applications including drug delivery, biotechnology and separation science.30 Among the many successful applications of MIPs in various areas, one of the most promising uses of MIPs has been in the area of drug delivery.31–34
Drug delivery is the practice of managing and administering a pharmaceutical compound to accomplish an optimal therapeutic effect. Drug delivery systems (DDS) have been abundantly studied and have seen rapid growth in the last few decades. The drug delivery strategy, in which a drug is delivered to its target, plays a vital and significant role in its efficacy. It is known that some drugs need to be administered over an extended period of time (controlled release) to achieve the maximum therapeutic effect for drugs that are rapidly metabolized and eliminated from the body after administration. In addition, a higher or lower amount of a drug may induce toxicity or negligible target effects.35 Hence, scientists have come up with integrated approaches, involving the combination of pharmaceutical science, polymer chemistry and molecular biology, to achieve greater benefits resulting from the synergistic effect of these fields.36 MIPs have demonstrated significant advancement in DDS. Some notable and interesting works on MIP based DDS were carried out by the collaborative efforts of Alvarez-Lorenzo, Puoci and their co-workers.37–40
In our previous published review on MIP based drug delivery vehicles, we provided a comprehensive discussion on various aspects of micro- and nano-scale systems for drug delivery, polymeric DDS and the advantages of MIP based drug delivery methods.36 This review is written as a continuation of the previous review and we intend to discuss the articles that have appeared from 2014 until now on MIP based DDS. It is worth mentioning that we will not provide the details of MIP based DDS fundamental features (i.e. the rationale of MIP use in DDS) as we have already discussed this comprehensively in our previous review.36 In the present review, we have included reports on targeted and stimuli responsive MIP systems due to the recent surge of interest on various types of DDS for anti-cancer drug release at specific locations such as cancerous cells (targeted delivery). On the other hand, MIP materials have also been successfully manipulated as stimuli responsive polymeric materials, which can be triggered by slight alterations in their environmental parameters. We have included all of these works detailing different therapeutic administration routes.
Application of MIP based drug delivery systems
Various applications of MIP based DDS will be discussed according to their therapeutic administration routes in the following sections.MIP applications in topical skin and transdermal routes
MIP based DDS have been exploited for both topical skin administration and transdermal routes. The former is generally defined as administration by applying the drug to body surfaces, such as the skin or mucous membranes, to treat ailments using a large range of dosage forms including creams, foams, gels, lotions, and ointments. In the latter route, active ingredients are delivered onto the skin for systemic distribution.Transdermal or skin routes have been used amply for drug introduction, owing to their many benefits, including enhanced bioavailability, prolonged duration of action resulting in a lower dosage frequency, diminished side effects, more uniform plasma levels and improved therapeutic effects due to the maintenance of plasma levels.41
Li et al.42 studied the controlled release of salicylic acid (SA), a beta-hydroxy acid and an anti-inflammatory drug known for its efficacy to reduce pain and diminish fever, and skin-conditioning agents using an MIP. Mild doses of SA have also been proven to be successful in topical skin applications for the treatment of seborrheic dermatitis, acne and psoriasis. However, it can cause severe side effects such as acute skin irritation, stinging and inflammation, together with moderate chemical burns if it is applied in high concentration. In their study, Li and co-workers synthesized two different MIPs and compared their performance. In the first approach, a sol–gel based MIP was prepared using 3-(aminopropyl)triethoxysilane (APTEOS) and trimethoxyphenylsilane as functional monomers, and tetraethyl orthosilicate (TEOS) as the cross-linker. In the second approach, an organic MIP was fabricated, where 1-(4-vinylphenyl)-3-(3,5-bis(trifluoromethyl)phenyl)urea was employed as a functional monomer. The sol–gel MIP showed the highest capacity and specificity for SA in an aqueous environment, unlike the organic MIP. The sol–gel MIP revealed a high specificity with an imprinting factor of 9 and demonstrated a binding capacity of 11.6 μg for SA per mg of polymer, that is, 1.2% SA, complying with dermatological formulations. The release of SA was predominantly diffusion controlled according to power law.
It has been shown in the literature that transdermal delivery of nicotine is beneficial as an aid to smoking cessation therapy, based on the hypothesis that a constant plasma level of nicotine reduces the craving for nicotine and therefore, aids in the abstinence from smoking. However, the development of nicotine transdermal patch materials faces many drawbacks, due to their poor physical withstanding properties and low permeation resistance. Thus, Ruela and co-workers43 administered nicotine via a transdermal method based on an MIP, due to its high lipid solubility and good permeation ability in the skin. The MIP particles were prepared in the presence of nicotine, methacrylic acid (MAA), and ethylene glycol dimetacrylate (EGDMA) via a bulk polymerization method. The resultant MIP materials were characterized successfully using Fourier transform infrared spectroscopy (FT-IR) and thermal analysis, as well as drug binding studies on the MIP. The nicotine MIP particles exhibited a suitable release rate in vitro, and the permeation capacity was also studied by directly anchoring MIP particles onto porcine ear skin in a vertical Franz-type glass diffusion cell apparatus with a magnetic stirrer. The skin permeation studies showed that the cumulative amount of nicotine that permeated was 655 ± 29 μg cm−2 at 24 h using the MIP particles, which was relatively close to the value of 709 ± 56 μg cm−2 at 24 h for the commercial patch, Nicopatch. It is worth noting that the total release was dependent on the types of vehicles used, such as polar and non-polar MIPs and NIPs (non-imprinted polymers) as shown in Fig. 1. For this purpose, propylene glycol (PEG) was selected as the polar (P) vehicle and mineral oil as the non-polar (NP) vehicle. It can be seen from Fig. 1A that no differences were found between the amounts of nicotine released at 32 h from the MIP-P and NIP-P delivery systems, whereas the amount of nicotine released at 32 h from the NIP-NP delivery system was higher than that of the MIP-NP delivery system (Fig. 1B), demonstrating that imprinting sites can hold nicotine for an increased amount of time in transdermal systems. The reason for this behavior lies in the vehicle polarity, and can be explained by the accumulation of hydrophilic drug near the surface of the hydrophobic based polymer carrier, which causes a high burst effect to be observed for both the MIP-P and MIP-NP delivery systems. However, non-polar vehicles favor molecular recognition mechanisms as they have similar polarity to porogen solvents allowing the three-dimensional conformation of the imprinted sites, thus mimicking the synthetic conditions. On the contrary, polar vehicles do not depict the same behavior, owing to selective interactions that decrease, due to modifications in the three-dimensional conformation of these imprinted sites. Furthermore, nitroglycerin (TNG) is considered a useful vasodilator drug, which is intensely employed as a nitrate donor agent to maintain certain cardiac conditions, including angina and congestive heart failure. However, this drug has very short life span (1–4 min) in liver and exhibits less than 1% bioavailability in the human metabolism system. Thus, to maintain a suitable concentration of nitrate and its bioavailability in the blood, transdermal routes using MIPs as DDS have been developed successfully in one report by Mohebali and his co-workers.44 The TNG imprinted nanospheres were fabricated by precipitation polymerization in the presence of MAA as a functional monomer, and trimethylolpropane trimethacrylate (TRIM) as a crosslinking agent. Significant reaction parameters, such as reaction medium, porogen solvents, cross-linker content, and initial monomer concentration, were investigated to achieve a highly efficient MIP material. The mean diameter of the TNG imprinted nanospheres for MIP was calculated to be 40 nm (ranging between 20 and 68 nm). It was found that the MIP prepared in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6:10 molar ratio of template, monomer, and cross-linker performed better in terms of its binding capacity, as well as its in vitro drug release profile in simulated biological fluid (phosphate buffer solution, PBS pH 7) via a dialysis bag method at 37 °C. However, the MIP material was not investigated using real transdermal studies.
:6:10 molar ratio of template, monomer, and cross-linker performed better in terms of its binding capacity, as well as its in vitro drug release profile in simulated biological fluid (phosphate buffer solution, PBS pH 7) via a dialysis bag method at 37 °C. However, the MIP material was not investigated using real transdermal studies.
 | ||
| Fig. 1 Release profile of nicotine from transdermal formulations containing MIP and NIP particles in different vehicles: (A) polar and (B) non-polar (reproduced with permission Elsevier publications from ref. 43). | ||
MIP application using ocular routes
Ocular delivery routes are a widely used method in DDS. However, serious obstacles including high lachrymal turnover and dynamics cause the quick precorneal removal of the drug, subsequently reducing its bioavailability and remedial response. Furthermore, a high frequency of drug use or an excessive dosage of drugs may pose potential risks to patients. Due to obstacles including low bioavailability and loading capacity of various ophthalmic solutions, ointments and gels, MIP based DDS may be the most fascinating area of research, as MIPs offer solutions for many similar problems. In general, various imprinted hydrogels used as soft contact lenses have been introduced in this category, where different compositions of these soft contact lens hydrogels exhibit different imprinting effects, drug loading capacities, and drug release. Alvarez-Lorenzo and co-workers carried out a few good studies in this area.45,46 For excellent binding and rebinding analysis, stable imprinted cavities are required, demanding strict control on the all the constituents, especially the cross-linking agent concentration. It has been reported that an increase in the amount of cross-linker may cause problems in optical transparency, flexibility and the swelling behavior of the lenses. It has been shown in many studies that a low concentration of cross-linker (55%) is more desirable in the preparation of soft contact imprinted lenses.47 Furthermore, extra care has to be taken to allow for water content while using HEMA as a backbone monomer, which is necessary for oxygen dissolution and diffusion into the surface of the cornea.48–50Brimonidine (BRN), an alpha 2 adrenergic agonist drug, is available as an ocular eye drop used to treat ocular hypertension. It is considered a safe and effective option for glaucoma, is well tolerated, and has few allergy concerns. In a recent study by Hediye et al.,51 BRN imprinted hydrogels were prepared using 2-hydroxyethyl methacrylate (HEMA) as a backbone monomer, MAA, methacrylamide (MAAM) and 4-vinylpyridine (4-VP) as the functional monomers and EGDMA as a cross-linker monomer. These imprinted hydrogels were studied for their performance in aqueous media. In this work, HEMA was utilized as a solvent too. The study showed that the selection of appropriate monomers and their content significantly influence the performance and specificity of the final MIP hydrogel. It was observed from the results that a mixture of functional monomers offered a higher loading capacity in optimized MIP hydrogel. Hence, poly(HEMA-co-MAA) showed superior binding properties compared to other copolymers. It was observed that all MIP hydrogels (prepared using any of the monomers) displayed superior affinity to BRN when compared to non-imprinted polymers (NIPs). The optimized MIP hydrogel with a BRN:MAA molar ratio of 1:8 exhibited an adequate amount of drug loading and controlled release, unlike MIP systems made using other molar combinations. The real analytical performance of the as-prepared systems was investigated in physiological saline (NaCl 0.9%) and artificial tear solution. From the Korsmeyer–Peppas equation, ‘n’ values of 1.1702 and 1.4702 were obtained in physiological saline and artificial tear solution, respectively, indicating that more than one model could be used to fit the drug release profile. Nevertheless, due to the high value of ‘n’ (>0.89), the super-case II transport release model was suggested, where a drug is released by both diffusion and relaxation of the polymer chains.
A series of poly(2-hydroxyethyl methacrylate) microspheres (PHEMA-MIPMs) and non-imprinted microspheres (NIPMs) were prepared via precipitation polymerization using gatifloxacin (GFLX), HEMA and EGDMA as the template molecule, functional monomer and cross-linker, respectively. The effects of the variation in the various components on the drug loading capacity, binding ability, and size of the microspheres with excellent flexibility were studied. The optimized study concluded that 1 vol% of HEMA and 70 mol% of EGDMA were the suitable initial monomer and cross-linker concentrations to prepare uniform and small sized PHEMA-MIPMs microspheres. The as-prepared PHEMA-MIPM microspheres showed a good sustained release and cumulative release of GFLX when compared to NIPMs, owing to the firmly embedded GFLX inside the imprinted cavities as recorded during the initial 48 h. This was primarily due to the photo-sensitivity and easy degradation of GFLX in solution over a long period of time.52
MIPs in oral therapeutic applications
Oral drug delivery routes are considered to be the most appropriate and persuasive route, despite some negative physiological effects of the gastrointestinal tract, such as irritation, metabolism, variation in delivery rates and interference due to the presence of food. Therefore, MIP materials have been employed considerably in oral therapeutic applications.In an interesting report, the Suedee group53 proposed temperature stimulated MIP nanoparticles selective for (R)-thalidomide. Firstly, a polymer MIP network was fabricated via both a covalent approach and a physical approach in the presence of (R)-thalidomide, a mixture of functional monomers including MAA, a fluorescently active 2,6-bis(acrylamido)pyridine and MBA. In the grafting procedure, the polymer is covalently attached, whereas a physical approach means that that polymer is just simply deposited onto the surface of already synthesized and functionalized acrylate poloxamer nanoparticles that are capable of thermo-stimulated responsiveness. The drug delivery capacity of the as-prepared MIP was assessed in cancer cells using green fluorescence measurements. It was observed that grafted MIP was capable of delivering the highest amount of the nontoxic (R)-thalidomide at the cancer tissue, after an appropriate shift in the temperature. In addition, the as-prepared recognition materials within the poloxamer nanoparticles were successfully employed as a promising anticancer agent to penetrate cells and release the drug, thereby initiating apoptosis in multidrug-resistant cells.
5-Fluorouracil (5-FU) is a prominent anticancer drug which is amply used in the treatment of several solid cancers. Due to its rapid metabolism in the body, continuous administration of 5-FU is required in order to maintain high serum concentrations, and maintain its therapeutic activity. This means that high dosages of this drug are required, which produces serious toxic effects.54 This can be avoided through the regulated release of 5-FU, as reported using many drug delivery devices composed of polysaccharides and polypeptides.55 However, it is believed that due to some of the excellent features of MIPs, including the controlled release of drugs with a narrow therapeutic index and their function as drug carrier vehicles, the over-concentration of drugs within body can be prevented, which subsequently inhibits adverse side effects. The use of MIPs increases bioavailability, and reduces side effects compared to some traditional delivery systems. To address the challenges and to improve the administration of 5-FU, many new procedures have been reported recently.
Hashemi-Moghaddam et al.56 fabricated magnetic nanoparticles coated with 5-FU imprinted polymers, for use in controlled drug release experiments for mouse breast cancer. The authors investigated several parameters, such as tumor-enlargement delay, tumor-doubling time inhibition ratio, and histopathology. The obtained results proved that the MIP suppressed tumor growth effectively in the presence of a magnetic field, unlike free 5-FU and the MIP without a magnetic field. Fig. 2 shows the results from the tumor growth inhibition ratio study, performed in different treatment groups and concentrations of 5-FU in various tissues in mice.
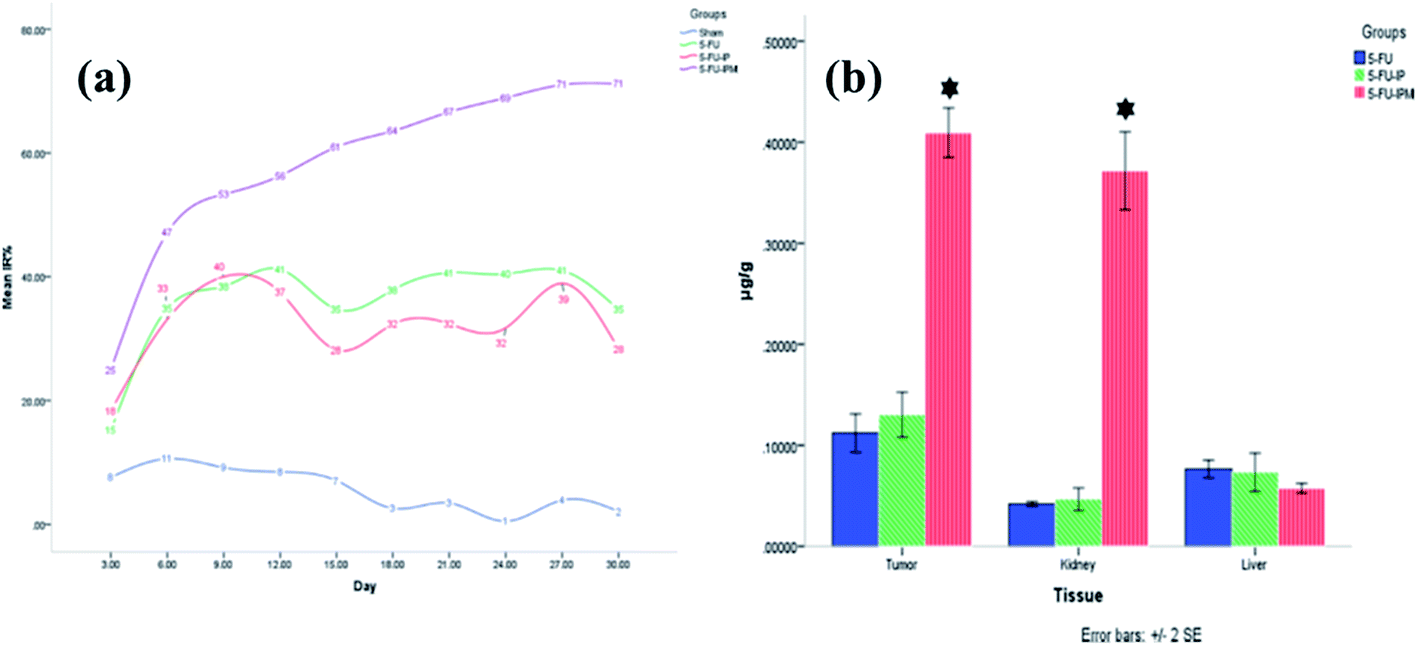 | ||
| Fig. 2 (a) Tumor growth inhibition ratio (IR%) of different treatment groups on selected days. 5-FU: 5-fluorouracil; 5-FU-IP: 5-fluorouracil imprinted polymer; 5-FU-IPM: 5-fluorouracil imprinted polymer with magnetic field. (b) Concentration of 5-FU in tumor, liver, and kidney tissues of tumor-bearing mice using different treatments. 5-FU: 5-fluorouracil; 5-FU-IP: 5-fluorouracil imprinted polymer; 5-FU-IPM: 5-fluorouracil imprinted polymer with magnetic field. * Indicates a significant difference compared with other groups (reproduced with permission from Elsevier Publications from ref. 56). | ||
In order to explore the potential of temperature and magnetic bi-responsive MIP systems (TMMIPs) in controlled drug delivery, Li et al.57 synthesized Fe3O4-encapsulating carbon nanospheres via free radical polymerization, followed by the grafting of N-isopropylacrylamide (NIPAM) as the temperature responsive monomer, in the presence of N,N′-methylene bisacrylamide (MBA) and 5-FU as the cross-linker and template, respectively. The as-synthesized TMMIPs were extensively and systematically investigated using field emission scanning electron microscopy (FE-SEM), FT-IR, thermogravimetry (TGA), UV-Visible spectrophotometry (UV-Vis), transmission electron microscopy (TEM), vibrating sample magnetometry (VSM), and dynamic light scattering (DLS). The schematic design, SEM and TEM characterization results are assembled in Fig. 3. The SEM and TEM analysis revealed that the TMMIPs were mono-disperse spherical shaped particles with an average diameter of 152 nm, possessing rough surfaces due to polymer grafting (as shown in the inset of Fig. 3B(a)). The data in Fig. 4a and b show the release rate of 5-FU from the TMMIPs and TMNIPs at 25 °C and the release rate of 5-FU from the TMMIPs at different temperatures, respectively. It is obvious that the thermo-stimulation (swelling and shrinking) of the TMMIPs is due to the incorporation of NIPAM, unlike the TMNIPs (non-imprinted polymer). It is also noteworthy that the release rate of 5-FU increased with increasing temperature and nearly 90.75% of drug was released at 45 °C. This phenomenon was attributed to the shrinking of the TMMIPs which induces hydrophobicity, resulting in weaker hydrogen bonding between the template and the functional monomer. On the other hand, when the temperature of the TMMIPs was below the lower critical solution temperature (LCST, i.e. 39.3 °C), the 5-FU release was impeded.
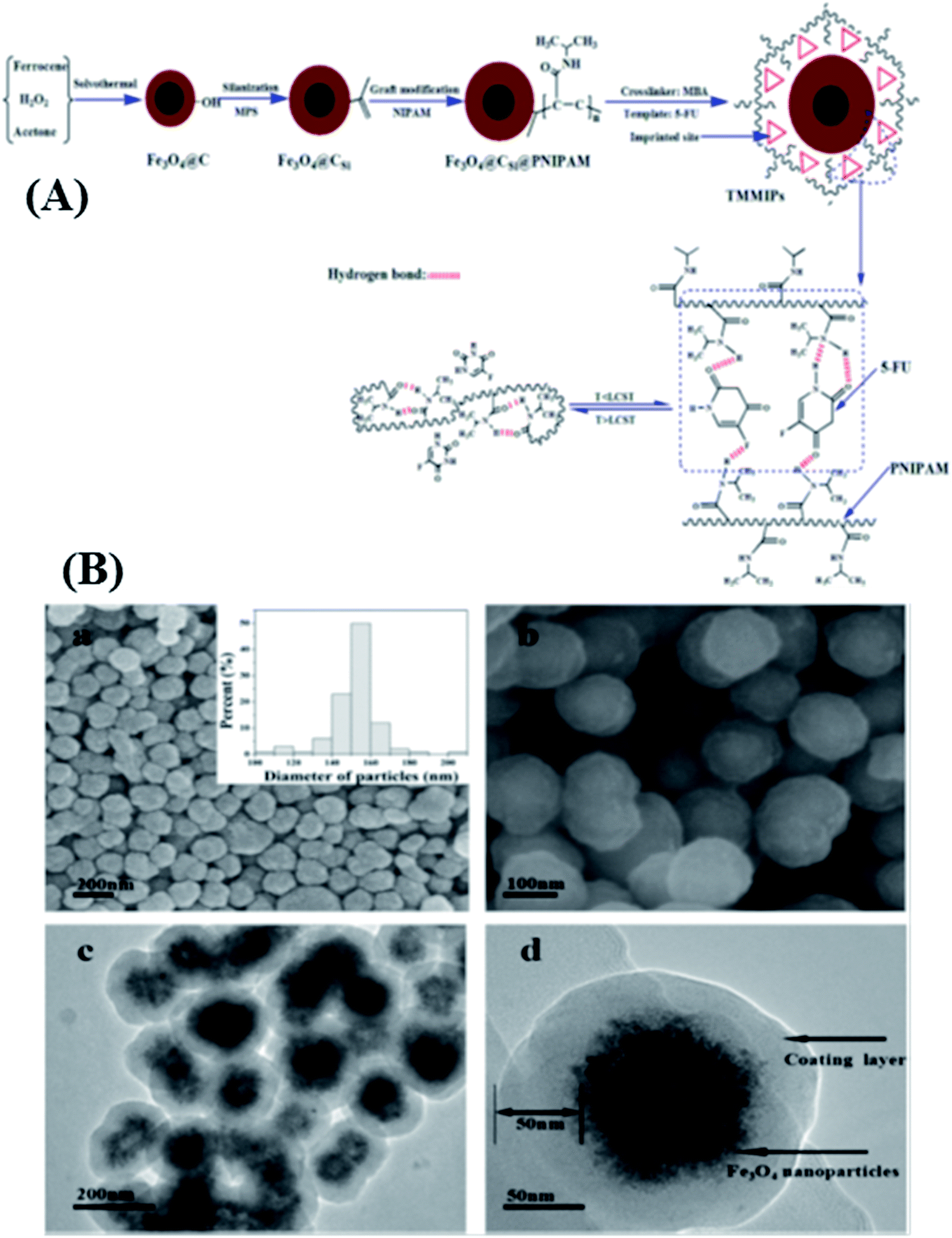 | ||
| Fig. 3 (A) Synthetic routes of TMMIPs and the reversible thermosensitive swelling/shrinking transition of TMMIPs. (B) FESEM (a and b) and TEM (c and d) images of TMMIPs; size distribution of TMMIPs (inset of (a)) (reproduced with permission from Elsevier Publications from ref. 57). | ||
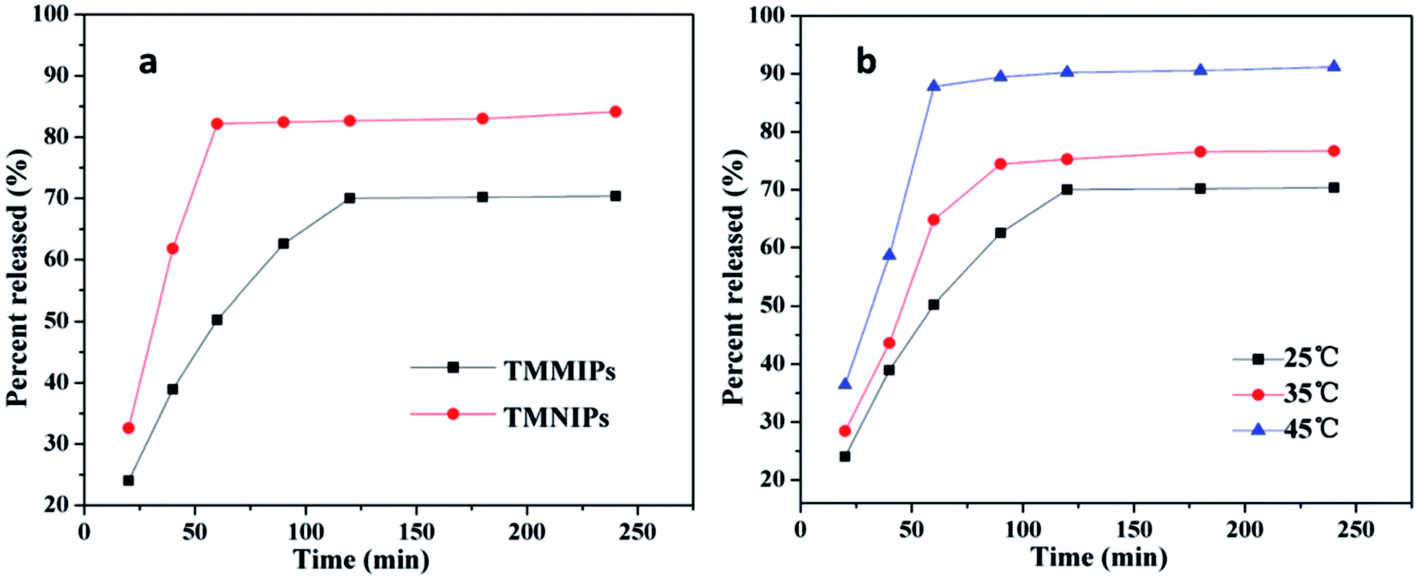 | ||
| Fig. 4 (a) Release rate of 5-FU from TMMIPs and TMNIPs at 25 °C; (b) release rate of 5-FU from TMMIPs at different temperatures (reproduced with permission from Elsevier Publications from ref. 57). | ||
Asadi et al.58 proposed a study on the controlled delivery of risperidone (RSP, 3-[2-[4-(6-fluoro-1,2-benzisoxazol-3-yl)-1-piperidinyl]ethyl]-6,7,8,9-tetrahydro-2-methyl-4H-pyrido[1,2-a]pyrimidin-4-one), an approved antipsychotic drug belonging to the chemical class of benzisoxazole derivatives used for the brain. It usually exhibits low bioavailability due to extensive first pass metabolism, and non-targeted delivery results in numerous side effects. The sustained and targeted delivery of this drug may effectively improve its bioavailability and achieve the desired drug concentration. RSP imprinted polymer nanoparticles were synthesized using MAA and TRIM via a mini emulsion polymerization, and were characterized using SEM. The results showed that RSP imprinted nanoparticles demonstrated superior binding capacity and a low release rate in 1% wt sodium dodecyl sulfate in aqueous solution as measured using high performance liquid chromatography with a UV detector. This significant difference in the release of imprinted nanoparticles and non-imprinted polymers could be attributed to the existence of specific sites in the imprinted nanoparticles that had a strong interaction with the RSP molecules.
In a similar protocol, paclitaxel (PTX), an anticancer drug, was loaded into imprinted nanoparticles and the release properties were studied.59 The MIPs were prepared by using PTX as a template, MAA and methyl methacrylate as non-covalent functional monomers, and EGDMA and TRIM as cross-linker agents. The reaction parameters, including the type of cross-linker agent employed and the template-monomer and monomer-cross-linker molar ratios, were tuned. FT-IR analysis revealed the successful interaction between PTX and the functional monomer in each MIP. In all the prepared MIPs, a burst release of PTX was observed at pH 5.0 during the initial stage of the release profile. However, the release rate gradually decreased and became constant for a period of 350 h. The initial burst release of PTX was ascribed to molecules which were adsorbed on the surface. Later, the release rate of PTX molecules became sustained and slow for a longer time period as they were tightly bound to the polymer matrix. The release of drug was attributed to diffusion and polymer degradation through a network of interconnected pores in the matrix of the polymer. It was also discussed that a higher amount of TRIM introduced higher cross-linking in the polymer, which was largely responsible for the slow release of PTX. The as-fabricated MIP nanoparticles provided a binding capacity that was 12 times higher compared to NIP and showed a very slow and controlled release of PTX.
Parisi et al.60 proposed an interesting approach where the drug controlled release ability of magnetic MIP nanoparticles for a 9H-carbazole derivative was exploited in cancer treatment. The MIP nanoparticles were composed of a template, MAA, EGDMA and magnetite. Later, the resulting materials were characterized using SEM and FT-IR analysis. The as-prepared 9H-carbazole imprinted materials displayed excellent selective recognition, controlled drug release, and a high magnetic response capacity during in vitro drug release and cytotoxicity studies on different cancer cell lines, such as HeLa and MCF-7. In the cytotoxicity studies, the controlled release of the 9H-carbazole derivative from the imprinted materials exhibited good inhibitory activity against the tested cancerous cells, which was initiated by apoptosis due to a dramatic growth arrest, compared to the controls. To improve the low bioavailability of curcumin, a versatile anti-inflammatory and anti-cancer agent, Piletska and co-workers61 prepared curcumin-imprinted magnetic nano-sized polymers (nanoMIPs) for the controlled delivery of curcumin. NanoMIPs were synthesized using various functional monomers including MAA, ethylene glycol methacrylate phosphate (EGMP), TRIM, HEMA and itaconic acid (ITA). These functional monomers were selected after calculating their binding energies via a leapfrog method using molecular modeling studies, based on the strong affinity between the template and monomers. The characterization study showed that the particle sizes of the as-prepared nanoMIPs were dependent on the UV irradiation time during polymer formation. The presence of specific monomers was shown to be significant in ensuring the effective binding of curcumin and important in determining the rate of release. The EGMP-based nanoMIPs demonstrated the highest binding affinity towards the template and the slowest controlled release of curcumin into the buffer solution, while the HEMA based polymer exhibited the weakest affinity for the template, and a rate of release that was significantly higher than that of EGMP. This was consistent with the results obtained using the Leapfrog algorithm. The batch studies showed that the EGMP-based nanoMIPs released 253 μg curcumin per particle, compared to the EGMP-based NIPs, which released only 129 μg per particle. On the other hand, 46.8 μg curcumin per particle per day (predicted time of curcumin release per days ∼ 5.4 days) was released from the EGMP-based nanoMIPs, compared to 105 μg curcumin per particle per day for the EGMP-based nanoNIPs (predicted time of curcumin release per days ∼ 1.2 days).
Erythromycin (ERY) is considered to be a broad antimicrobial spectrum drug and is negligibly susceptible to causing allergic reactions. It is employed as an important remedy for respiratory tract, genital, skin, and soft tissue infections. The effective administration of ERY faces various challenges such as degradation in gastric acid, low absorption, harmful effects after extended administration, and the lack of novel therapeutic approaches and dosage courses. In order to address these problems, Kempe et al.62 successfully synthesized ERY imprinted nanoparticles based on non-covalent bonding, using MAA, TRIM, 2,2′-azobisisobutyronitrile (AIBN), and acetonitrile via precipitation polymerization. These nanoparticles were capable of sustained drug release under physiological conditions. The obtained Langmuir binding isotherms demonstrated the excellent binding capacity (87 μmol g−1), drug loading efficiency (∼87%) and loading capacity (76 mg of ERY per g nanocarriers) of the ERY-MIP nanocarriers, as compared to ERY-NIP nanocarriers (44 μmol g−1), after incubation in PBS for 20 h. Furthermore, the ERY-MIP nanocarriers demonstrated an outstanding delivery of ERY for over a week, after which time roughly 82% (∼62 mg g−1) of the initially loaded ERY had been discharged. The burst release followed the zero-order model, whereas the Korsmeyer–Peppas model provided a better fit for the sustained release of the drug with an ‘n’ value determined to be 0.79, indicating non-Fickian diffusion.
For an effective oral colon-specific DDS, the administration of a drug must be controlled, prolonged, and colon targeted, to avoid degradation in the gastrointestinal area. Hence, Men et al.63 synthesized metronidazole (MTZ) molecular surface imprinted microspheres (MIP-PSSS/CPVA), using cross-linked polyvinyl alcohol microspheres (CPVA) with sodium 4-styrene sulfonate as the functional monomer, and MBA, followed by a cerium salt–hydroxyl group redox initiation system. The as-prepared MIPs showed a greater binding capacity of up to 115 mg g−1 for MTZ at pH 1.0. The in vitro release of the MTZ-loaded MIP-PSSS/CPVA was found to be greatly affected by the pH of the buffer. The release behavior revealed that a sustained release of MTZ was observed for 26 h in the simulated colon fluid (pH 7.4), whereas a negligible release and an abrupt release of MTZ were observed in the simulated gastric fluid (pH 1.0) and in the simulated small intestinal fluid (pH 6.8), respectively.
Doxorubicin (DOX) is an amply used anticancer drug which often requires a high drug loading amount and controlled release rate in order to achieve better results. Wang et al.64 exploited an MIP technique for imprinting DOX biocompatible hollow microcapsules (MIMs) composed of carboxymethyl cellulose-(chitosan/alginate). The as-prepared MIMs were capable of encapsulating and delivering DOX via their sufficiently thin imprinted shells, which function as the gate or channel, while DOX functions as the stopper. The binding or release of drug leads to the closing/opening of the gate and thus controls the transfusion of drug through the mesochannels of the imprinted microcapsules. The gate/channel opening was found to be dependent on the interaction of the carboxyl groups of O-carboxymethyl chitosan, which in turn was determined by the lower pH value. The drug loading capacity of the MIMs was calculated to be 155.1 mmol g−1. After its burst release within the first 12 h, the release of DOX was sustained for over 168 h, due to the imprinted sites being blocked by DOX molecules, which entered the imprinted sites through electrostatic interaction. A very long time was required for the acid solution to overcome the electrostatic interaction of the DOX molecules with their specific sites.
Lopez et al.65 studied the controlled release of nafcillin (NAF), using biocompatible dummy templates such as (+)-6-aminopenicillenic acid (APA), or ampicillin ((+)-6-aminobenzylpenicillin)sodium salt (AMP) MIP nanospheres, prepared via a sol–gel route using TEOS and APTEOS as precursors. The in vitro release studies of the drug were examined under simulated gastrointestinal tract conditions. The study showed that the combination of these materials provide better results, resulting in the release of NAF and APA imprinted nanospheres typically after a latency-period of 24 hours, whereas the non-imprinted and the AMP-imprinted material showed a burst release during the first 5 h.
Tryptophan (Trp) is one of the 20 essential amino acids that cannot be produced by our body. Therefore, the only way get Trp is through the consumption of a Trp rich diet. It is also necessary for the production of serotonin and melatonin, essential for many important functions in the human body. Based on these facts, there is always a need for the facile, precise, fast, and economic method for the controlled release of Trp. Guney and Serin66 demonstrated a stimuli-responsive Trp imprinted hybrid polymer sol–gel as a promising method for controlled release. Prior to obtaining the MIP, a coupling reaction of 3-isocyanatopropyltriethoxysilane with the hydroxyl end groups of poly(propylene glycol) (PPG) was carried out to obtain trimethoxysilane terminated poly(propylene glycol) (PPG-ICPTS). This was used to prepare MIP in the presence of APTEOS, tetramethoxysilane and methyltrimethoxysilane (MTMOS). The release of Trp molecules was dependent on the swelling behavior of the various polymer materials containing different constituents (i.e. different functional monomers and amounts), in the presence of various ratios of solvent mixtures, due to the porous polymer network. A mixture of 75% ethanol and 25% water was found optimum to obtain the maximum swelling degree, as observed by pseudo-Fickian diffusion calculated from different swelling rates. Furthermore, the polymer gel prepared using MTMOS as a precursor displayed the maximum release of Trp at swelling equilibrium depending on the pH. The release rate of Trp was measured to be slightly faster at low pH, owing to the weak interactions between Trp and APTEOS, in comparison with in a high pH buffer.
Tang et al.67 focused on the preparation of MIPs using a molecular crowding approach with polystyrene (PS) as a macromolecule and aminoglutethimide (AG) as the drug, via free-radical precipitation polymerization, in order to achieve a zero-order extended release of AG. The application of the molecular crowding approach was derived from the notion that biological macromolecules evolve and function within highly crowded/dense intracellular or extracellular environments. Therefore this approach can profoundly influence and improve thermodynamic activities and biological processes by several orders of magnitude, through volume exclusion effects that reduce diffusion rates and enhance the binding rates of macromolecules. Therefore, the MIPs were prepared using PS in the presence of MAA and EDMA in an optimized ratio, as functional monomer and cross-linker, respectively. Other functional monomers such as AM (acrylamide) and 4-VP were also tested, resulting in either burst release or the loss of the controlled release ability of the MIPs. Similar findings were obtained when TRIM and divinylbenzene (DVB) were used to prepare MIPs. The Brunauer–Emmett–Teller (BET) surface area and pore volumes of the crowding-assisted MIPs were nearly 10 times higher than those of the controlled MIPs (without the crowding conditions). The crowding-assisted MIPs exhibited an imprinting factor of 6.02, while this was 1.19 for the controlled MIPs, as measured from equilibrium adsorption experiments. The release profile of AG continued for nearly 18 h using the MIPs, in comparison to 10 h in the case of the controlled MIPs, and both materials exhibited zero-order behavior. In contrast, using the other two types of controlled particles, the AG molecules were quickly released in the first hour (∼85%). This study also successfully examined the relative bioavailability of crowded-assisted MIPs and NIPs in commercial AG tablets with values of 266.3% and 57.7%, respectively.
Azithromycin (AZM) is the first semi-synthetic macrolide antibiotic, which is derived from erythromycin and belongs to the azalide group. It shows low bioavailability of approximately 37%, which is usually attributed to oral administration and the variation of drug concentration in the body. Hence, to improve its therapeutic value, Sheybani et al.68 used mesoporous MIP nanoparticles as a sustained release system prepared via precipitation polymerization using poly(MAA-co-EGDMA) nanoparticles. As expected, the AZM-MIP nanoparticles demonstrated a higher binding capacity than the control NIP nanoparticles. The Korsmeyer–Peppas model, which is used to analyze the drug release behavior from a polymeric matrix, determined the value of n (diffusion exponent) to be 0.59, indicating a non-Fickian diffusion mechanism where both the diffusion of drug and polymer swelling control the rate of drug release. Furthermore, it was observed that the release data of the drug was fitted well using the Korsmeyer–Peppas model (R2 = 0.9669), and this fit was better than those attempted using other models. The drug release plots showed an initial burst release, followed by a release of nearly 24% of loaded AZM within first day. During the next two days, approximately 14% of the AZM was released from the MIP nanoparticles and after a week, 78% of the drug was released. Moreover, the cytotoxicity results depicted good non-toxicity to the L929 cells line, along with biocompatibility.
Kaamyabi et al.69 reported multifunctional activity, including pH responsiveness, thermoresponsiveness and high drug loading capacity, in a DOX imprinted polymer composite [poly(NIPAAM@Fe3O4MNPs/TMSPMC/DOX)] synthesized by the radical polymerization of methacrylate functionalized Fe3O4 nanoparticles (3-(trimethoxysilyl)propyl methacrylate, TMSPMC) with NIPAM and DOX in the presence of AIBN and EGDMA as the polymerization initiator and cross-linker, respectively. The MIP was characterized using FT-IR, SEM, X-ray powder diffraction analysis (XRD), UV-Vis, VSM, TGA and differential thermal analysis. The as-proposed DOX polymer was prepared by using a solution with a LCST of 40 °C, which can be beneficial in cancer drug delivery, since the temperature of cancer cells is higher than normal ones. Secondly, the MIP showed a response at a more acidic pH than the pH value of normal cells (pH = 5.8). Thus, DOX molecules were effectively released selectively in the affected area (about 88% in cancer cells and only 12% in normal cell), as tested in a simulated body fluid (SBF, pH = 7.4). Thirdly, the DOX imprinted polymer was grafted onto the surface of nanoparticles, allowing for a high number of imprinting sites. The pH and temperature based drug release profiles are shown in Fig. 5A and B, respectively, and support the claims made in the study.
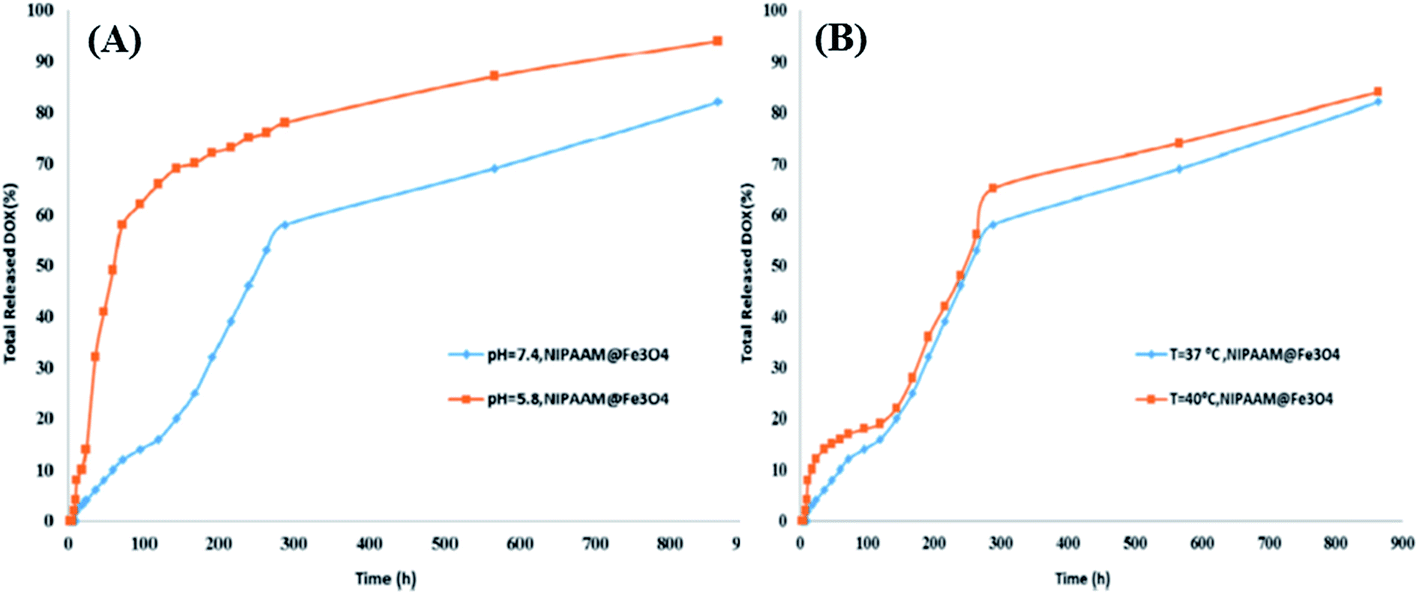 | ||
| Fig. 5 (A) DOX release from poly(NIPAAM@Fe3O4MNPs/TMSPMC/DOX) in different pHs, and (B) DOX release from poly(NIPAAM@Fe3O4MNPs/TMSPMC/DOX) in different time intervals and temperatures (reproduced with permission from Elsevier Publications from ref. 69). | ||
Poor solubility in water and instability in physiological media restrict oral delivery routes for quercetin (QC), a valuable anti-inflammatory, antioxidant and anticancer agent. For a better oral delivery route, Hemmati et al.70 studied a highly selective and efficient magnetic molecularly imprinted polymer (MMIP) nanogel with a core–shell structure, synthesized by a sol–gel process composed of QC, Tragacanth Gum (TG) as cross-linker, Fe3O4/SiO2 nanoparticles, and an N-vinyl imidazole (VI) functional monomer. Their efficiencies were compared with a magnetic non-imprinted polymer (MNIP). As expected, the MMIP showed superior properties compared to the MNIP in terms of swelling, and followed a Langmuir model to provide rapid equilibrium behavior. The in vitro release of the drug was studied as a function of time and pH of the media. It was observed that about 30% of the total loaded QC was released from the MMIP in the first 8 h, owing to a weakened degree of protonation of the nitrogen and oxygen atoms of VI and QC, resulting in low electrostatic interaction between them, while the MNIP released about 73% of adsorbed QC in the same time at pH 7.0. However, at pH 2.0, the ratio of the released amount of QC in the first 8 h increased up to 80% and 40% for the MNIP and MMIP, respectively. The increased release of QC is attributed to the intensified protonation of QC molecules and VI macromolecules, and the electrostatic attraction becoming restricted due to the electronic repulsion of the protonated groups. This observation revealed that the synthesized MMIP is pH-sensitive. The in vitro drug release mechanism revealed a value of 0.45 for ‘n’ using the Korsmeyer–Peppas equation, suggesting that the release of QC followed a pure diffusion mechanism (Fickian), by the usual molecular diffusion of the drug due to a chemical potential gradient.
Zheng and co-workers71 studied the controlled release of L-tyrosine by synthesizing a molecular imprinted membrane (MIM) using L-tyrosine, CS–GEL (chitosan–gelatin) and sulfuric acid, as a template molecule, a functional monomer and a cross-linker, respectively. The as-synthesized MIM was characterized by SEM, UV-Vis, XRD and FT-IR and was utilized in vitro successfully. It was found that only 58.6% of L-tyrosine was released from the MIM in the first 2 h at 37 °C, while the percentage of the L-tyrosine released from the non-imprinted membrane was 90.6% in the initial 1.5 h.
Donepezil, a piperidine derivative drug, is administrated for treatment of Alzheimer disease. This drug has poor solubility in water, hence lipid based-formulations (LBFs) may be employed to improve its oral administration. Recently, in an interesting and detailed study proposed by Ruela et al.,72 donepezil imprinted microparticles were prepared to be incorporated in an LBF for the sustained release of donepezil, with the aim of controlled release to optimize the safety and efficacy of this drug after oral administration. Two imprinted polymers were prepared using different constituents. The first MIP (MIP-1) was obtained using a template, MAA and HEMA as functional monomers, and EDMA as a cross-linker in the presence of porogen solvents and a radical initiator. The preparation of the second MIP (MIP-2) was carried out in 2 steps. In the first step, a template was mixed with MAA, EDMA and the radical initiator in the presence of solvents, followed by the addition of EDMA, HEMA and d glycidyl methacrylate (GMA). The addition of hydrophilic monomers (HEMA and GMA) in the second MIP protocol was performed in order to overcome the limitations of non-specific hydrophobic interactions between the template and MIPs in an aqueous environment. The corresponding NIPs were prepared in a similar way, but without a template. To probe the in vitro release of donepezil, the as-prepared polymer particles were incorporated into different LBF vehicles, such as oleic acid (OA), OA:C (oleic acid containing Cremophor), and OA:GMO (oleic acid:monoolein), maintaining different drug and polymer ratios. It was observed that there was no difference between the drug release from MIP-1 and NIP-1 in the absence of an oil vehicle. For MIP-1, the donepezil was completely released from the oleic acid (control without copolymers) at 60 min, indicating that the oleic acid did not have a significant effect on the sustained release of donepezil. The systems with drug:polymer ratios of 1:3 and 1:6 exhibited similar multiphasic release profiles, where burst release was observed for few minutes, followed by a second phase in which most of the drug molecules were released, suggesting a zero order kinetic model, which is characterized by gradual diffusion. In the last phase, the remaining drug was released. This multiphasic release kinetic profile was attributed to the heterogeneous binding site distribution in the MIPs. When the ratio of drug:polymer was decreased to 1:18, the donepezil release rate was significantly reduced. The study of MIP-2 (drug:polymer ratio of 1:18) showed a biphasic drug release behavior. It was suggested that MIP-2 offered an increased rate of donepezil release than MIP-1 due to enhanced wettability caused by more hydroxyl groups, as a result of the epoxide ring opening reaction of the GMA. Moreover, in both MIPs, Fickian diffusion of the drug from the polymer matrix was suggested based on the Korsmeyer–Peppas model.
MIP in intravenous routes
To avoid drug degradation, and improve drug bioavailability, MIP-based systems have also been administrated intravenously in in vitro testing.Olanzapine belongs to the second generation of antipsychotic drugs and is administrated for Schizophrenia. However, the oral administration of this drug is not very beneficial, as roughly 40% of the drug is degraded before reaching systemic circulation, limiting its clinical application. On the other hand, higher dosage frequencies may lead to many adverse effects such as tremors, dry mouth, weight gain, somnolence and so on. Thus, Asadi and co-workers73 applied “fructose”, which performed a dual function as a monomer and as a biodegradable cross-linker, for the synthesis of an olanzapine magnetic fluorescent multi-core shell structured MIP via co-precipitation polymerization, for brain targeted prolonged and controlled release. When studying the effect of pH on the extraction of drug, it was found that the preferable pH value was 6.8. This excellent approach exploited the dual function of the MIPs, whose magnetic properties can promote their easy localization near affected brain tissues, while the degraded fructose produced during drug release can be consumed by brain cells. In this work, both in vitro as well as in vivo (performed on rat brain via intravenous injection) experiments were carried out for the drug release studies, with acceptable results except for good cell compatibility and non-toxicity of the fabricated magnetic MIP nanocomposites.
Asadi et al.74 developed an effective nanodevice using new fluorescent multi-core–shell structured magnetic MIP nanoparticles based on biodegradable materials (tannic acid, a high molecular weight polyphenol used as a cross-linker), for the targeted, sustained and controlled release of 5-FU. The nanoparticles were synthesized via a mini-emulsion polymerization technique as shown in Fig. 6. The structure and surface morphology of the as-synthesized samples were studied by various techniques. The release of 5-FU was found to rely upon pH, and the drug was immediately released within 2 h in simulated gastric fluid (pH 1.2). However, it showed a sustained release for up to 120 h in plasma simulating fluid (pH 7.4). The in vivo assay was carried out in a rat model, by injecting MIP solution intravenously, and the in vivo fluorescent images confirmed that the MIP drug carriers were effectively administrated into the liver as guided by an external magnetic field. These studies demonstrated the efficacy of the drug carrier for cancer treatment, as in vitro and in vivo measurements were performed on human breast tumor cells with satisfactory anti-cancer properties and biocompatibility.
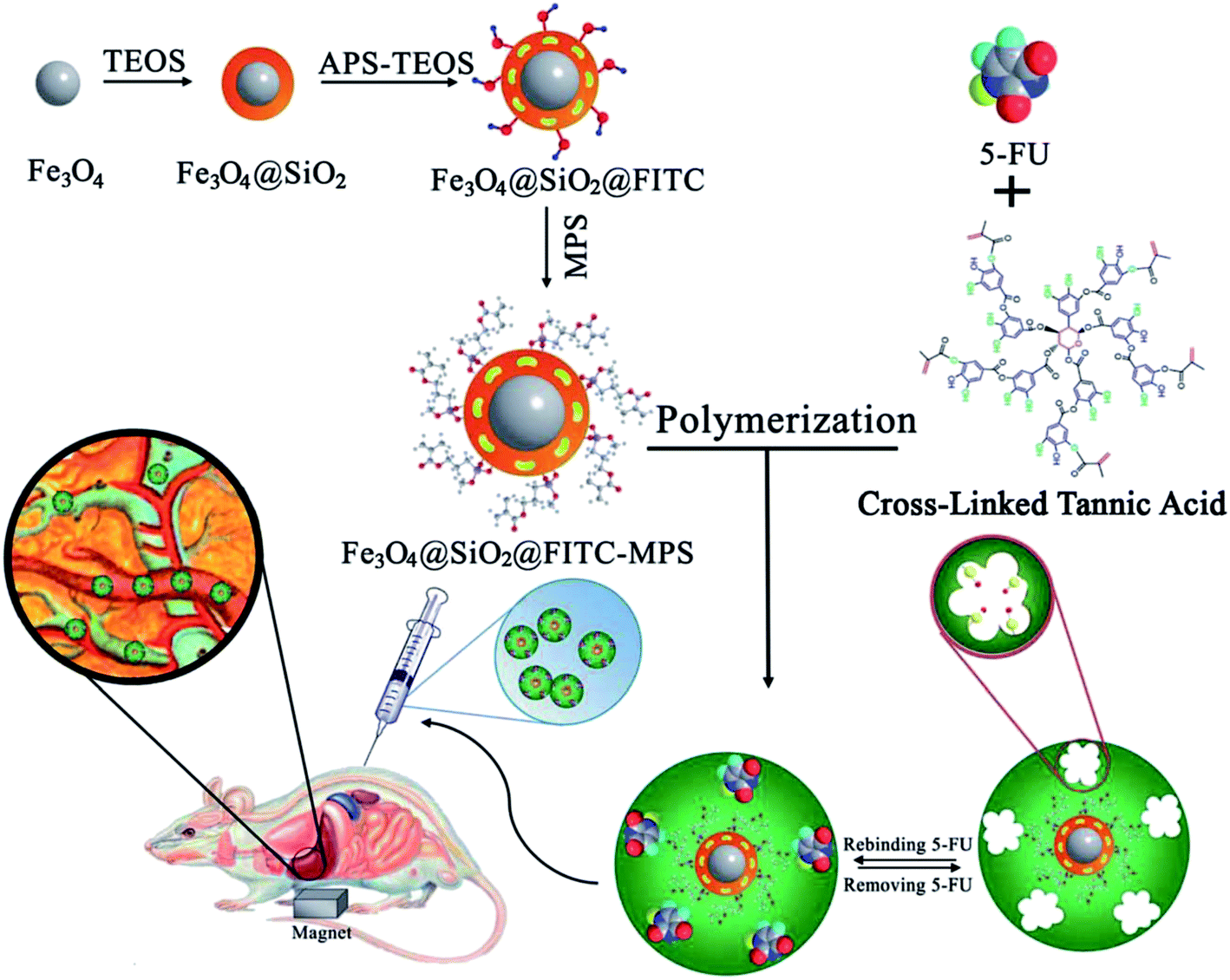 | ||
| Fig. 6 Schematic representation of the preparation and characterization of magnetic molecularly imprinted polymer (MMIP) nanoparticles as a 5-FU carrier (TEOS: tetraethyl orthosilicate, APS: 3-aminopropyl trimethoxysilane, FITC: fluorescein isothiocyanate, 5-FU: 5-fluorouracil, MPS: methacryloxypropyl trimethoxysilane) (reproduced with permission from Royal Society of Chemistry from ref. 74). | ||
Miscellaneous applications of MIP
In one report, the enantioselective capability of MIPs was exploited and its drug releasing behavior was examined in detail as a proof-of-concept study. Kupai et al.75 proposed an excellent strategy for the expansion of enantioselective controlled drug release systems, where imprinted polymers were synthesized in the presence of 1-(1-naphthyl)ethylamine hydrogen perchlorate as the template and some optically active macrocycles as functional monomer anchors. The MIPs were applied to enantioselective recognition and pH-sensitive drug delivery. In order to study the effect of ring size, chirality of the macrocycle and the presence of the pyridine subunit, three crown ether type functional monomers and the corresponding precyclization analogues were synthesized and used to fabricate the MIPs. Among the various fabricated MIPs based on the different constituents, the MIP based on pyridino-18-crown-6 ether (S,S)-5 as a functional monomer was selected for all further studies, owing to its favorable C2 symmetry and allyloxy substituent. The results revealed that the release of the R-enantiomer was dependent on pH, while the S-enantiomer delivery was prompt irrespective of pH for MIP-1 (composition: (R)-1/(S,S)-5/EDMA; stoichiometry: 1/1/40). This was in contrast to MIP-2 (composition: rac-1/(S,S)-5/EDMA; stoichiometry: 1/1/40), which did not show enantioselective potential. Furthermore, the pore size and surface area were other influencing factors in the drug release. The in vitro release kinetic data obtained for the majority of both the imprinted and control polymers were best fitted using a first order model, which demonstrated an immediate mode of drug release. The data also indicated that the rate limiting step for the release was diffusion, which was not strongly concentration dependent. The drug release data showed that the MIPs provided instantaneous drug release at basic pH due to a gradual increase in the deprotonation ability of 1-(1-naphthyl)ethylamine hydrogen perchlorate.Present hurdles in MIP based DDS
In the last 2–3 years, many interesting studies have appeared that show excellent potential for the advancement of MIP based DDS. After reviewing the above-mentioned articles, it is very clear that plenty of studies have been focused on targeted drug delivery for anti-cancer drugs. New research has been attempting to address the challenges that come up during the development of MIP based DDS such as controlling the balance between pharmacokinetics and pharmacodynamics, the toxicity and biocompatibility of the employed polymer, selective recognition, efficacy of drug loading and release, and the behavior of the MIPs in the surrounding environment. As obvious from the results of MIP based studies, loading capacities as well as drug release are the biggest problems that occur during the fabrication and analysis of MIPs. It has been discussed in some reports that drug loading is often not sufficient, which leads to low and ineffective drug release. Moreover, sometimes drug loading is adequate, but either the quick release of all the drug molecules or an initial burst release of a high percentage of the drug molecules cause many severe side effects, such as high dosage or influences on normal cells. Nonetheless, it should be kept in mind that a moderate burst release may be beneficial to provide an initial higher dose at the start of an antibiotic drug therapy. Thus, release profiles may vary according to the types of drug administration. Another highly important aspect of MIP based DDS is the formation of imprinted cavities in MIP gels prepared by using water soluble monomers that are not quite as densely cross-linked and may suffer due to swelling behavior under external conditions. Another obstacle arises from low bioavailability, which obviously opens the door for new administration routes and the fabrication of polymeric materials, which in turn requires a broad range of monomer and cross-linker materials.As discussed in our previous review, there is a great demand for polymerization reactions that can be carried out in an aqueous environment, as this is required for in vitro mimicking unlike reactions performed in organic solvents. Thus, careful optimization and tuning of experimental parameters and constituents are necessary. In order to improve drug efficacy, the distribution of the binding sites and morphological uniformity are essential. Hence, better polymerization procedures need to be utilized. Furthermore, synthesis reproducibility is also a vital factor for ensuring the robustness and practicability of MIPs. It was also noticed from studying the literature that very few articles showed in vivo studies. It should be essential step in all research to include an in vivo study, and only then, the real application of MIPs in DDS can be compared to other competitive polymeric materials. There is also a need for peptide or protein imprinted DDS in order to extend the applications of MIPs. These kinds of applications are scarce.
What is more puzzling is that the intended route of drug administration was not even discussed in several papers. This makes the research less fruitful for readers.
It is also worth mentioning that MIP based drug carriers have not been employed in all drug delivery routes. They have only been applied in ocular and transdermal routes (in vivo tests) and oral (but only in vitro tests). They have not been tested, for instance, for vaginal drugs. Therefore, this field also needs due attention in future.
Hence, it can be deduced that MIPs as drug delivery devices have not yet found any successful application. There is still a lot of research that needs to be carried out to overcome some of the problems stated above. However, interesting investigations and research are in progress to apply MIPs in many forms of DDS.
Conclusions and future perspective
It is obvious after reviewing the articles on MIPs, that it is a well-established area of research with great potential in diverse applications, such as sensing, enantioselective separation and molecular recognition, solid phase extraction and environmental analysis. However, their use as active biomedical devices, such as in drug delivery vehicles, is still in the nascent stages of development as discussed in our work. Although MIP-based drug carriers have been employed in a variety of drug delivery routes, their biocompatibility and efficacy of drug release have posed great challenges among scientists.Although the development and application of MIPs in drug delivery are still in their infancy, constant and steady efforts of researchers will steer the research with increasing innovative ideas, and new intelligent drug delivery systems with commercial value can be expected in the near future. However, it can be expected, on the basis of current emerging trends and procedures, that more and more stimulating future progress in drug delivery and therapeutic monitoring will depend on real-time analysis, to give rise to an intelligent, feedback responsive outcome for MIP-based DDS systems. There is a need to direct efforts to develop solid oral dosage forms consisting of MIPs for therapeutic protein and peptide delivery and the targeted release of potent drugs that address life threatening disease such as cancer. This could be achieved via epitope imprinting techniques and merging pharmaceutical and bio-engineering skills.
Based on present multidisciplinary investigations into polymeric material synthesis, soon MIP-based DDS systems may be a real contender for drug delivery vehicle materials in biomedical devices. Furthermore, several fancy and hybrid studies have established the potential of implantable microchip-based controlled drug delivery. DDS based on a hybrid of MIPs and microchips may prove to be promising biomedical devices, and may make these future prospects into reality.
Acknowledgements
The financial support of the Kwangwoon University fund in 2016 is greatly acknowledged.References
- M. V. Polyakov, Zh. Fiz. Khim., 1931, 2, 799–805 Search PubMed
.
- G. Wulff and A. Sarhan, Angew. Chem., 1972, 84, 364 CrossRef
- R. Arshady and K. Mosbach, Macromol. Chem., 1981, 182, 687–692 CrossRef CAS
- S. A. Zaidi, Anal. Methods, 2015, 7, 7406–7415 RSC
- S. Li, X. Huang, M. Zheng, W. Li and K. Tong, Sensors, 2008, 8, 2854–2864 CrossRef
- G. M. Murray, A. L. Jenkins, A. Bzhelyansky and O. M. Uy, Johns Hopkins APL Tech. Dig., 1997, 18, 464–472 CAS
- X. Zhang, M. Zhu and S. Li, J. Inorg. Organomet. Polym. Mater., 2014, 24, 890–897 CrossRef CAS
- J. Wackerlig and R. Schirhag, Anal. Chem., 2016, 88, 250–261 CrossRef PubMed
- L. Chen, X. Wang, W. Lu, X. Wu and J. Li, Chem. Soc. Rev., 2016, 45, 2137–2211 RSC
- M. Subat, A. S. Borovik and B. J. Konig, J. Am. Chem. Soc., 2004, 126, 3185–3190 CrossRef CAS PubMed
- E. Byrne, E. Oral, J. Z. Hilt and N. A. Peppas, Polym. Adv. Technol., 2002, 13, 798–816 CrossRef
- J. Z. Hilt, M. E. Byrne and N. A. Peppas, Chem. Mater., 2006, 18, 5869–5875 CrossRef CAS
- A. Nematollahzadeh, W. Sun, C. S. A. Aureliano, D. Lütkemeyer, J. Stute, M. J. Abdekhodaie, A. Shojaei and B. Sellergren, Angew. Chem., Int. Ed., 2011, 50, 495–498 CrossRef CAS PubMed
- W. Chen, W. Lei, M. Xue, F. Xue, Z. H. Meng, W. B. Zhang, F. Qu and K. J. Shea, J. Mater. Chem. A, 2014, 20, 7165–7169 Search PubMed
- W. J. Cheong, S. A. Zaidi and Y. S. Kim, Bull. Korean Chem. Soc., 2014, 35, 3115–3118 CrossRef CAS
- S. A. Zaidi, Electrophoresis, 2013, 34, 1375–1382 CrossRef CAS PubMed
- S. A. Zaidi and W. J. Cheong, J. Sep. Sci., 2008, 31, 2962–2970 CrossRef CAS PubMed
- S. A. Zaidi and W. J. Cheong, J. Chromatogr. A, 2009, 1216, 2947–2952 CrossRef CAS PubMed
- S. A. Zaidi, K. M. Han, S. S. Kim, D. G. Hwang and W. J. Cheong, J. Sep. Sci., 2009, 32, 996–1001 CrossRef CAS PubMed
- R. Jang, K. H. Kim, S. A. Zaidi, W. J. Cheong and M. H. Moon, Electrophoresis, 2011, 32, 2167–2173 CrossRef CAS PubMed
- S. A. Zaidi, S. M. Lee and W. J. Cheong, J. Chromatogr. A, 2011, 1218, 1291–1299 CrossRef CAS PubMed
- A. Bhaskarapillai, N. Sevilimedu and B. Sellergren, Ind. Eng. Chem. Res., 2009, 48, 3730–3737 CrossRef CAS
- J. Wang, M. Xue, Z. Meng, Z. Xu and J. Luo, Anal. Methods, 2016, 8, 4413–4420 RSC
- X. Su, X. Li, J. Li, M. Liu, F. Lei, X. Tan, P. Li and W. Luo, Food Chem., 2015, 171, 292–297 CrossRef CAS PubMed
- S. A. Zaidi, Int. J. Electrochem. Sci., 2013, 8, 9936–9955 CAS
- S. A. Zaidi and J. H. Shin, Int. J. Electrochem. Sci., 2014, 9, 4598–4616 Search PubMed
- R. Suedee, C. Jantarat, W. Lindner, H. Viernstein, S. Songkro and T. Srichana, J. Controlled Release, 2010, 142, 122–131 CrossRef CAS PubMed
- H. Hiratani, A. Fujiwara, Y. Tamiya, Y. Mizutami and C. Alvarez-Lorenzo, Biomaterials, 2005, 26, 1293–1298 CrossRef CAS PubMed
- A. L. Hillberg, K. R. Brain and C. J. Allender, Adv. Drug Delivery Rev., 2005, 57, 1875–1889 CAS
- W. Chen, Y. Ma, J. Pan, Z. Meng, G. Pan and B. Sellergren, Polymers, 2015, 7, 1689–1715 CrossRef CAS
- P. Lulinski, Acta Pol. Pharm., 2013, 70, 601–609 CAS
- R. Schirhag, Anal. Chem., 2014, 86, 250–261 CrossRef PubMed
- E. Oral and N. A. Peppas, Proceed. Intern. Pharm. Technol. Symp., 2000, 10, 59–60 Search PubMed
- M. Byrne, K. Park and N. A. Peppas, Adv. Drug Delivery Rev., 2002, 54, 149–161 CrossRef CAS PubMed
- C. Alvarez-Lorenzo and A. Concheiro, Chem. Commun., 2014, 50, 7743–7765 RSC
- S. A. Zaidi, Drug Delivery, 2014, 1–10, DOI:10.3109/10717544.2014.970297
- F. Puoci, F. Iemma and N. Picci, Curr. Drug Delivery, 2008, 5, 85–96 CrossRef CAS
- C. Alvarez-Lorenzo and A. Concheiro, From drug dosage forms to intelligent drug delivery systems: a change of paradigm, in Smart materials for drug delivery, ed. C. Alvarez-Lorenzo and A. Concheiro, RSC, Cambridge, 2013, vol. 1, ch. 1, pp. 1–32 Search PubMed
- Molecularly imprinted polymers as components of drug delivery systems, in Handbook of molecularly imprinted polymers, ed. C. Alvarez-Lorenzo and A. Concheiro, Smithers Rapra Publishing, Shrewsbury, UK, 2013, ch. 8, pp. 309–349 Search PubMed
- C. Alvarez-Lorenzo, C. Gonzalez-Chomon and A. Concheiro, Molecularly imprinted hydrogels for affinity-controlled and stimuli responsive drug delivery, in Smart materials for drug delivery, ed. C. Alvarez-Lorenzo, A. Concheiro, H. J. Schneider and M. Shahinpoor, RSC, Cambridge, 2013, vol. 1, ch. 21, pp. 228–260 Search PubMed
- A. Ahad, M. Aqil and A. Ali, Int. J. Biol. Macromol., 2014, 64, 144–149 CrossRef CAS PubMed
- B. Li, J. Xu, A. J. Hall, K. Haupt and B. T. S. Bui, J. Mol. Recognit., 2014, 27, 559–565 CrossRef CAS PubMed
- A. L. M. Ruela, E. C. Figueiredo and G. R. Pereira, Chem. Eng. J., 2014, 248, 1–8 CrossRef CAS
- A. Mohebali, M. Abdoussa, S. Mazinani and P. Zahedi, Polym. Adv. Technol., 2016, 27, 1164–1171 CrossRef CAS
- C. Alvarez-Lorenzo, H. Hiratani, J. L. Gomez-Amoza, R. Martínez-Pacheco, C. Souto and A. Concheiro, J. Pharm. Sci., 2002, 91, 2182–2192 CrossRef CAS PubMed
- C. Alvarez-Lorenzo, F. Yanez, R. Barreiro-Iglesias and A. Concheiro, J. Controlled Release, 2006, 113, 236–244 CrossRef CAS PubMed
- C. J. White, A. Tieppo and M. E. Byrne, J. Drug Delivery Sci. Technol., 2011, 21, 369–384 CrossRef CAS
- H. Hiratani, Y. Mizutani and C. Alvarez-Lorenzo, Macromol. Biosci., 2005, 5, 728–733 CrossRef CAS PubMed
- H. Hiratani and C. Alvarez-Lorenzo, Biomaterials, 2004, 25, 1105–1113 CrossRef CAS PubMed
- F. Tashakori-Sabzevar and S. A. Mohajeri, Drug Dev. Ind. Pharm., 2015, 41, 703–713 CrossRef CAS PubMed
- M. O. Hediye, A. S. T. Sayyed, K. Reza, S. R. Maryam and A. M. Seyed, Current Drug Development, 2015, 12, 717–725 CrossRef
- X.-F. Lu, Y.-F. Shi, H.-L. Lv, Y.-Y. Fu, D. Ma and W. Xue, J. Mater. Sci.: Mater. Med., 2014, 25, 1461–1469 CrossRef CAS PubMed
- A. Suksuwan, L. Lomlim, T. Rungrotmongkol, T. Nakpheng, F. L. Dickert and R. Suedee, J. Appl. Polym. Sci., 2015, 41930, 1–21 Search PubMed
- K. R. Johnson, K. K. Young and W. Fan, Clin. Cancer Res., 1999, 5, 2559–2565 CAS
- E. Fournier, C. Passirani, N. Colin, P. Breton, S. Sagodira and J. P. Benoit, Eur. J. Pharm. Biopharm., 2004, 57, 189–197 CrossRef CAS PubMed
- H. Hashemi-Moghaddam, S. Kazemi-Bagsangani, M. Jamili and S. Zavareh, Int. J. Pharm., 2016, 476, 228–238 CrossRef PubMed
- L. Li, L. Chen, H. Zhang, Y. Yang, X. Liu and Y. Chen, Mater. Sci. Eng., C, 2016, 61, 158–168 CrossRef CAS PubMed
- E. Asadi, S. Azodi-Deilami, M. Abdouss, D. Kordestani, A. Rahimi and S. Asadi, Korean J. Chem. Eng., 2014, 31, 1028–1035 CrossRef CAS
- F. A. Ishkuh, M. Javanbakht, M. Esfandyari-Manesh, R. Dinarvand and F. Atyabi, J. Mater. Sci., 2014, 49, 6343–6352 CrossRef CAS
- O. L. Parisi, C. Morelli, F. Puoci, C. Saturnino, A. Caruso, D. Sisci, G. E. Trombino, N. Piccia and M. S. Sinicropi, J. Mater. Chem. B, 2014, 2, 6619–6625 RSC
- E. V. Piletska, B. H. Abd, A. S. Krakowiak, A. Parmar, D. L. Pink, K. S. Wall, L. Wharton, E. Moczko, M. J. Whitcombe, K. Karim and S. A. Piletsky, Analyst, 2015, 140, 3113–3120 RSC
- H. Kempe, A. P. Pujolras and M. Kempe, Pharm. Res., 2015, 32, 375–388 CrossRef CAS PubMed
- J. Men, B. Gao, L. Yao and Y. Zhang, J. Macromol. Sci., Part A: Pure Appl.Chem., 2014, 51, 914–923 CrossRef CAS
- P. Wang, A. Zhang, Y. Jin, Q. Zhang, L. Zhang, Y. Peng and S. Du, RSC Adv., 2014, 4, 26063–26073 RSC
- T. D. Lopez, M. A. Francos, A. F. Gonzalez, M. E. Diaz-Garcia and R. Badia-Laino, Curr. Top. Med. Chem., 2015, 15, 262–270 CrossRef PubMed
- O. Guney and E. Serin, J. Appl. Polym. Sci., 2016, 42913, 1–8 Search PubMed
- L. Tang, C. Y. Zhao, X. H. Wang, R. S. Li, J. R. Yang, Y. P. Huang and Z. S. Liu, Int. J. Pharm., 2015, 496, 822–833 CrossRef CAS PubMed
- S. Sheybani, T. Hosseinifar, M. Abdouss and S. Mazinanib, RSC Adv., 2015, 5, 98880–98891 RSC
- S. Kaamyabi, D. Habibi and M. M. Amini, Bioorg. Med. Chem. Lett., 2016, 26, 2349–2354 CrossRef CAS PubMed
- K. Hemmati, A. Masoumi and M. Ghaemy, Carbohydr. Polym., 2016, 136, 630–640 CrossRef CAS PubMed
- X. F. Zheng, Q. Lian, H. Wu, H. Liu and S. Song, Russ. J. Appl. Chem., 2015, 88, 160–168 CrossRef CAS
- A. L. M. Ruela, E. C. de Figueiredo, M. B. de Araújo, F. C. Carvalho and G. R. Pereira, Eur. J. Pharm. Sci., 2016, 93, 114–122 CrossRef CAS PubMed
- E. Asadi, M. Abdouss, R. M. Leblanc, N. Ezzati, J. N. Wilson and D. Kordestani, Polymer, 2016, 97, 226–237 CrossRef CAS
- E. Asadi, M. Abdouss, R. M. Leblanc, N. Ezzati, J. N. Wilson and S. Azodi-Deilami, RSC Adv., 2016, 6, 37308–37318 RSC
- J. Kupai, E. Rojik, P. Huszthy and G. Szekely, ACS Appl. Mater. Interfaces, 2015, 7, 9516–9525 CAS
| This journal is © The Royal Society of Chemistry 2016 |