
Dual enzymatic dynamic kinetic resolution by Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase and Candida antarctica lipase B†
Ibrahim Karumea,
Musa M. Musa *a,
Odey Bsharata,
Masateru Takahashib,
Samir M. Hamdanb and
Bassam El Alia
*a,
Odey Bsharata,
Masateru Takahashib,
Samir M. Hamdanb and
Bassam El Alia
aDepartment of Chemistry, King Fahd University of Petroleum and Minerals, Dhahran, 31261, Saudi Arabia. E-mail: musam@kfupm.edu.sa; Fax: +966-13-860-4277; Tel: +966-13-860-7343
bDivision of Biological and Environmental Sciences and Engineering, King Abdullah University of Science and Technology, Thuwal 23955-6900, Saudi Arabia
First published on 4th October 2016
Abstract
The immobilization of Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase (TeSADH) using sol–gel method enables its use to racemize enantiopure alcohols in organic media. Here, we report the racemization of enantiopure phenyl-ring-containing secondary alcohols using xerogel-immobilized W110A TeSADH in hexane rather than the aqueous medium required by the enzyme. We further showed that this racemization approach in organic solvent was compatible with Candida antarctica lipase B (CALB)-catalyzed kinetic resolution. This compatibility, therefore, allowed a dual enzymatic dynamic kinetic resolution of racemic alcohols using CALB-catalyzed kinetic resolution and W110A TeSADH-catalyzed racemization of phenyl-ring-containing alcohols.
Introduction
The vital role of optically active compounds in pharmaceuticals, agrochemicals, and other synthetic chemicals unceasingly demands cheap and environmentally benign methodologies to obtain enantiomerically pure compounds. For example, drug efficacy could be influenced in cases where one enantiomer outrages the activity of the opposite enantiomer.1 Deracemization approaches that involve the formation of a single enantiomer from its corresponding racemate, such as cyclic deracemization, enantioconvergence, stereoinversion and dynamic kinetic resolution (DKR), are ideal alternatives to kinetic resolution (KR).2,3 DKR that couples a KR reaction with an in situ racemization for the slow-reacting enantiomer in KR has been the most advanced deracemization approach that exceeds the 50% yield limit of KR and reaches up to a 100% yield.3 Non-enzymatic systems, for example, chiral ferrocene–ruthenium complexes, have been employed in DKR of racemic alcohols.4,5 A significantly popular approach for DKR of alcohols relies on combining an enzymatic KR with a transition metal complex based racemization.2b,3a Core–shell beta-silicalite-1 micro composites were also used as racemization catalysts for secondary alcohols in a one-pot DKR with lipase-catalyzed KR.6Candida antarctica lipase B (CALB), known as Novozyme 435, is the most popular lipase used in the KR-catalyzed transesterification of alcohols.7 Lipases catalyze the enantioselective transesterification with preference to R-configured alcohols in KR, nevertheless W104A CALB mutant and proteases such as subtilisin have shown selectivity for S-alcohols in KR of secondary alcohols.8 The inexpensive and environmentally benign nature of enzymes presents a preferable alternative to challenges by organometallic catalysts. The scarcity of racemases due to the high stereoselectivity of the biosynthetic pathways, however, leaves less hope for a full enzymatic DKR approach. Alcohol dehydrogenases (ADHs), which catalyze the enantioselective redox reactions of ketones and their corresponding alcohols,9–11 have been successfully reported to accomplish racemization of enantiopure alcohols.12 In this front, we have shown that Thermoanaerobacter ethanolicus secondary alcohol dehydrogenase (TeSADH) mutants can be used to racemize enantiopure secondary alcohols.13,12a
TeSADH is a nicotinamide adenine dinucleotide phosphate (NADP+)-dependent ADH that is well-known for its high thermal stability and high tolerance to organic media.14 Xerogel-encapsulated W110A TeSADH mutant has been successfully employed in the asymmetric reduction of ketones in non-aqueous media.15 Unlike the wild-type TeSADH, W110A TeSADH reduces a variety of ketones with high conversions and enantiomeric excess (ee).15 Although W110A TeSADH follows Prelog's rule,16 the stereoselective reduction of prochiral ketones by TeSADH may occur by delivering the hydride from the undesired Si face producing the anti-Prelog alcohols, a phenomenon known as “selectivity mistakes”, leading to racemization (Scheme 1).12a Such selectivity mistakes may occur because of the reverse fit of substrates in the enzyme's active site.
 | ||
| Scheme 1 Racemization of enantiopure alcohols using W110A TeSADH. ADPR = adenosine diphosphoribose, R1 is more sterically hindered than R2. | ||
Herein, we successfully report a strategy and its implementation for a dual enzymatic DKR of secondary alcohols. This approach relies on racemization of enantiopure secondary alcohols using xerogel-immobilized W110A TeSADH in water-immiscible organic solvents, which enabled its use with CALB-catalyzed KR in one pot (Scheme 2).
 | ||
| Scheme 2 One-pot dual enzymatic DKR of secondary alcohols. CALB: Candida antarctica lipase B, R1 has higher Cahn–Ingold–Prelog priority that R2. | ||
Results and discussion
We started by immobilizing W110A TeSADH together with NADP+ and NADPH using the sol–gel method as described previously.15 The wet sol–gel (hydrogel) was then dried to produce a xerogel. We selected W110A TeSADH because it showed good performance in racemization of enantiopure alcohols and high tolerance to organic solvents.12a,15 We then conducted the racemization of (S)-1-phenyl-2-propanol [(S)-1a] using xerogel-immobilized W110A TeSADH in different organic solvents (Table 1). This substrate was selected because it was reported as a substrate for W110A TeSADH17 and its counterpart enantiomer as a substrate for CALB.18 Racemization of (S)-1a in water-miscible organic solvents like methanol and dimethyl sulfoxide (DMSO) showed no activity, which could be due to removal of water molecules required to maintain the enzyme's activity. The efficiency of W110A TeSADH-catalyzed racemization showed an improvement in various solvents with high log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P. Among the solvents tested, hexane and cyclohexane showed the best racemization performance (Table 1, entries 6 and 7). We also tested the racemization of (S)-1a using xerogel-immobilized W110A TeSADH in Tris–HCl buffer solution, and noticed an improvement in racemization efficiency. This indicates that using hydrophobic solvents like hexane restricts the diffusion of the substrate through the xerogel. The xerogel contains water that is enough to maintain the enzyme's active conformer and perhaps too little to trigger CALB-hydrolysis of ester products.15
P. Among the solvents tested, hexane and cyclohexane showed the best racemization performance (Table 1, entries 6 and 7). We also tested the racemization of (S)-1a using xerogel-immobilized W110A TeSADH in Tris–HCl buffer solution, and noticed an improvement in racemization efficiency. This indicates that using hydrophobic solvents like hexane restricts the diffusion of the substrate through the xerogel. The xerogel contains water that is enough to maintain the enzyme's active conformer and perhaps too little to trigger CALB-hydrolysis of ester products.15
|
||||
|---|---|---|---|---|
| Entry | Solvent | logPb |
% ee S enantiomerc | |
| Before | after | |||
| a Unless stated, reactions were performed at 50 °C and 200 rpm for 24 h using (S)-1-phenyl-2-propanol (0.04 mmol), xerogel [W110A TeSADH (0.2 mg), NADPH (1.0 mg) and NADP+ (1.0 mg)] in a solvent (1.0 mL).b logP (partition coefficient) values based on octanol–water partition coefficient.19c The ee of the corresponding acetate ester derivative of the produced alcohol was determined by chiral GC.d 0.6 mg of W110A TeSADH was used instead of 0.2 mg.e Aqueous medium [Tris–HCl (pH 8.0, 50 mM)/acetonitrile (95:5 v/v)]. |
||||
| 1 | DMSO | −1.4 | >99 | >99 |
| 2 | Methanol | −0.7 | >99 | >99 |
| 3 | MTBE | 0.9 | >99 | 96 |
| 4 | Dichloromethane | 1.3 | >99 | >99 |
| 5 | Toluene | 2.7 | >99 | 94 |
| 6 | Cyclohexane | 3.2 | >99 | 84 |
| 7 | Hexane | 3.8 | >99 | 81 |
| 8 | Hexane | 3.8 | >99 | 63d |
| 9 | Heptane | 4.3 | >99 | 96 |
| 10 | 2,2,4-Trimethylpentane | 4.4 | >99 | 93 |
| 11 | Octane | 4.8 | >99 | 92 |
| 12 | Tris–HCle | — | >99 | 59 |

The course of the racemization reaction of (S)-1a was monitored with time using free and immobilized W110A TeSADH to check the effect of the reaction medium and the enzyme immobilization on the racemization efficiency. We monitored the racemization of (S)-1a at various time intervals using free W110A TeSADH in Tris–HCl buffer solution (pH 8.0, 50 mM) and in biphasic system containing Tris–HCl buffer solution (pH 8.0, 50 mM)/hexane (1:1, v/v), and compared their performances with that for xerogel-immobilized W110A TeSADH in hexane (Fig. 1). Nearly complete racemization was observed after 20 h when the free enzyme was used in an aqueous medium (from >99% to 5% ee). Moderate racemization efficiency was noticed in a biphasic system containing Tris–HCl buffer solution/hexane (1:1, v/v) and these conditions resulted in a reduction in the ee of (S)-1a from >99% to 33% in 24 h. A rapid racemization was observed in the first 4 hours in Tris–HCl (54% ee) and in the biphasic system (64% ee). Good enzyme activity in both systems is attributed not only to the abundance of water required to maintain enzyme's activity but also to the free state of the enzyme, which facilitate the diffusion of the substrate in and out of the active site of the enzyme.
 | ||
| Fig. 1 Comparison of racemization of (S)-1a using free and immobilized W110A TeSADH in aqueous, biphasic and organic media. | ||
Racemization of (S)-1a using xerogel-immobilized W110A TeSADH containing 0.2 mg of the enzyme in hexane (1.0 mL) resulted in poor racemization efficiency, however, higher enzyme loading resulted in significant improvement (81% vs. 63% ee using 0.2 mg and 0.6 mg of the enzyme, respectively, Table 1). In a previously reported asymmetric reduction of phenyl-ring-containing ketones using xerogel-immobilized W110A TeSADH in hexane,17 isopropanol was used as a cosubstrate and a cosolvent, which might have facilitated the diffusion of the substrate to the active site, and thus resulting in high conversions to alcohols. The low racemization performance of the xerogel-immobilized W110A TeSADH in hexane when compared to that of the free enzyme is attributed to the slow diffusion rate of the substrate through the sol–gel matrix due to the absence of the cosubstrate. However, using immobilized ADHs in non-aqueous media to perform racemization of enantiopure alcohols is still interesting because of the versatile applications in such media.20
Xerogel containing W110A TeSADH (0.6 mg) and hexane as a solvent were adopted as optimum conditions for racemization of other enantiopure alcohols in non-aqueous media (Table 2). Under these conditions, racemization of (R)-1a gave better results than (S)-1a (Table 2, entries 1 and 2). Similar results were obtained in racemization of (S)-4-phenyl-2-butanol [(S)-1b] and (R)-4-phenyl-2-butanol [(R)-1b], however, with lower efficiencies (Table 2, entries 3 and 4). This is attributed to the structure of 1b, which seems to properly fit in the active site of W110A TeSADH and thus increasing the substrates' stereospecificity of the enzyme, and hence slowing racemization. TeSADH is reported to have two pockets within the active site, which are varying in size.21 The relatively small size of 1a might enable the entire molecule to fit in the large pocket with either direction allowing more selectivity mistakes hence the enhanced racemization. The improved racemization efficiency of (R)-alcohols compared to (S)-alcohols is attributed to the fact that W110A TeSADH obeys Prelog's rule, and thus expected to produce (S)-configured alcohols from the generated ketones whilst the (R)-configured alcohol is formed by selectivity mistakes; similar results were observed previously in W110A TeSADH-catalyzed racemization of phenyl-ring-containing enantiopure alcohols using free enzyme in an aqueous medium.12a Least racemization efficiency was observed for (R)-1-phenyl-2-butanol [(R)-1c] (98% to 90% ee), which is due to the extended chain length that might lead to a highly restricted fit of this enantiomer in the active site (i.e., high preference to (S)-enantiomer with less selectivity mistakes). Similar results were previously obtained using various mutants of TeSADH in racemization of (R)-1c using a free enzyme.13
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | % eeb | |
| before | after | ||||
| a Unless stated, reactions were performed at 50 °C at 200 rpm, for 24 h using the alcohol (0.04 mmol), xerogel-[W110A TeSADH (0.6 mg), NADPH (1.0 mg) and NADP+ (1.0 mg)] in hexane (1.0 mL).b The ee of the corresponding ester derivative of the produced alcohol was determined by chiral GC. | |||||
| 1 | (S)-1a | C6H5CH2 | CH3 | >99(S) | 63(S) |
| 2 | (R)-1a | C6H5CH2 | CH3 | >99(R) | 48(R) |
| 3 | (S)-1b | C6H5CH2CH2 | CH3 | >99(S) | 93(S) |
| 4 | (R)-1b | C6H5CH2CH2 | CH3 | >99(R) | 80(R) |
| 5 | (R)-1c | C6H5CH2 | CH3CH2 | 98(R) | 90(R) |

The ability of xerogel-immobilized W110A TeSADH to partially racemize enantiopure alcohols in hexane as the sole reaction medium encouraged us to attempt a one-pot dual enzymatic DKR that combines a CALB-catalyzed KR and W110A TeSADH-catalyzed racemization. The first requirement for a successful DKR is an efficient KR. We started by comparing the selectivity of CALB-catalyzed KR of (rac)-1a using selected acyl donors in hexane (Table 3). Isopropenyl acetate showed high activity (48% conv.) with moderate selectivity (E = 13–17). Similar results were obtained with vinyl acetate with lower selectivity. Methyl methoxy acetate showed excellent selectivity (E > 100) with low conversion (Table 3, entry 4). 2,2,2-Trifluoroethyl butyrate gave moderate conversion and selectivity (Table 3, entry 5).
|
||||
|---|---|---|---|---|
| Entry | Acyl donor | Conv. (%) | eepb (%) | E(R/S)d |
| a Unless stated, reactions were performed using (rac)-1a (0.05 mmol), acyl donor (0.1 mmol) and CALB (1.0 mg) in hexane (1.0 mL) at 50 °C at 50 rpm for 3 h.b The ee of the produced ester was determined by chiral GC.c Reaction after 6 h.d E values were calculated according to eqn (1) in the Experimental section. | ||||
| 1 | Isopropenyl acetate | 26 | 81(R) | 13 |
| 2 | Isopropenyl acetate | 48 | 78(R)c | 17 |
| 3 | Vinyl acetate | 31 | 75(R) | 10 |
| 4 | Methyl methoxy acetate | 5 | >99(R) | >100 |
| 5 | 2,2,2-Trifluoroethyl butyrate | 15 | 87(R) | 17 |

Compatibility of a stereoselective KR reaction with an efficient racemization method is an essential requirement for a successful DKR. We, therefore, proceeded to check compatibility of W110A TeSADH-catalyzed racemization and CALB-catalyzed KR in one-pot. The presence of water in xerogel might retard the CALB-catalyzed KR of alcohols. As previously reported,15 the xerogel contains more than 50% by weight water, and thus the two catalyst systems (i.e., xerogel-immobilized W110A TeSADH and CALB) were positioned such that direct contact is avoided as explained in the experimental section (see Fig. S2 of ESI†). Deracemization of (rac)-1a via DKR using CALB and W110A TeSADH dual enzymatic approach resulted in 74% conversion to the corresponding (R)-acetate ester in 68% ee. The calculated total composition of R enantiomers (i.e., (R)-1a and its corresponding (R)-acetate ester) was 62%, which indicates that CALB-catalyzed KR and W110A TeSADH are compatible (Fig. 2).
 | ||
| Fig. 2 GC chromatogram of products of DKR of (rac)-1a using CALB-catalyzed KR and W110A TeSADH-catalyzed racemization. | ||
Among the tested substrates, best DKR results were obtained with (rac)-1a (74% conv. & 68% ee, Table 4, entry 1); this is attributed to the good extent of racemization of its enantiopure forms by W110A TeSADH when compared with other substrates. Under the same conditions, (rac)-1b, (rac)-4-phenyl-3-butyn-2-ol [(rac)-1d] and (rac)-4-(4′-methoxyphenyl)-2-butanol [(rac)-1e], gave similar results (69–71% conv. & 58–59% total R), Table 4, entries 2, 4 and 5. Lowest activity was observed with (rac)-1c (15% conv. & 52% R) due to slow KR (Table 4, entry 3). To obtain an efficient DKR of alcohols, the rate of the racemization of the slow reacting enantiomer in KR should be faster than the rate of the esterification of the fast reacting alcohol.22 This is to ensure having a racemic alcohol over the course of the reaction and thus minimizing the selectivity mistakes that could be encountered in KR. This is apparently not the case for our racemization approach using xerogel-immobilized W110A TeSADH. Further investigation of various types of sol–gel with functionalized silanes that might improve substrate diffusion rate is needed; this might improve the racemization efficiency.
|
||||||
|---|---|---|---|---|---|---|
| Entry | Substrate | R1 | R2 | Conv. (%) | eeb (%) | Total Rc (%) |
| a Unless stated, reactions were performed using (rac)-1a–e (0.05 mmol), 2b (0.1 mmol), xerogel-immobilized enzyme [W110A (0.6 mg), NADP+ (1.0 mg) and NADPH (1 mg)], CALB (1 mg) in hexane (1.0 mL) at 50 °C at 50 rpm for 24 h.b The ee of the ester product was determined by chiral GC.c Total R (%) = R-alcohol + R-ester. | ||||||
| 1 | (rac)-1a | C6H5CH2 | CH3 | 74 | 68(R) | 62 |
| 2 | (rac)-1b | C6H5CH2CH2 | CH3 | 70 | 66(R) | 58 |
| 3 | (rac)-1c | C6H5CH2 | CH3CH2 | 15 | 99(R) | 52 |
| 4 | (rac)-1d | C6H5CC | CH3 | 71 | 63(R) | 58 |
| 5 | (rac)-1e | p-MeO-C6H4CH2CH2 | CH3 | 69 | 71(R) | 59 |
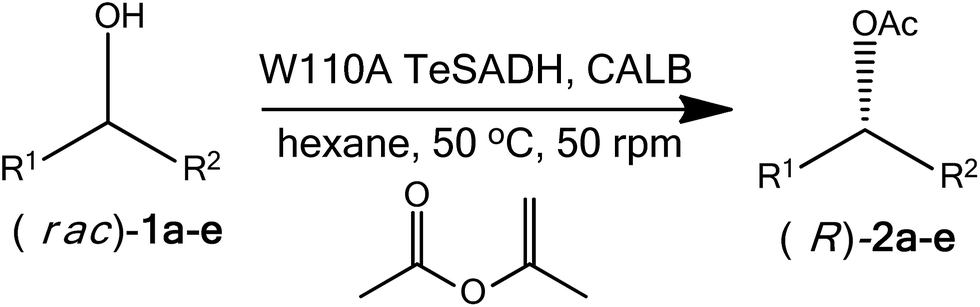
To our knowledge, this is the first report that shows that a dual enzymatic DKR of isolated alcohols is possible. Developing new mutants of TeSADH is crucial for the improvement of the racemization efficiency of enantiopure alcohols, and thus improving the effectiveness of the reported dual DKR. Extensive effort is currently conducted in our laboratory to generate new mutants of TeSADH through protein engineering to improve the racemization efficiency of TeSADH.
Conclusion
W110A TeSADH has shown activity in racemization of enantiopure alcohols in organic media contra to aqueous media required by the enzyme. We encapsulated W110A TeSADH using the sol–gel method and used the immobilized enzyme in organic solvents to racemize enantiopure phenyl-ring-containing alcohols. The use of W110A TeSADH-catalyzed racemization in pure hexane allowed the incorporation of this racemization method with in situ CALB-catalyzed KR. The results obtained in this work indicate that the two enzymes are compatible presenting an opportunity for one-pot dual enzymatic DKR. In a follow-up, we are currently conducting an extensive protein engineering studies for TeSADH to improve its racemization efficiency and investigating a variety of sol–gel prepared using different silanes with hydrophobic moieties that might facilitate substrate diffusion during racemization hence improving this DKR approach.Experimental
General
Compounds (R)-1a, (S)-1a, (R)-1b, (S)-1b, (rac)-1a, (rac)-1b, NADPH, NADP+, CALB (Novozyme 435), and solvents (HPLC grade) were used as purchased from Sigma-Aldrich; (R)-1c was obtained by CALB-catalyzed kinetic resolution of (rac)-1c as reported;23 (rac)-1c–e were synthesized by NaBH4 reduction of the corresponding commercially available ketones, as reported.24Capillary gas chromatography (GC) measurements were performed on Agilent 7890A gas chromatograph equipped with a flame ionization detector and an Agilent HP-Chiral-20B column (30 m, inner diameter 0.32 mm, film thickness 0.25 μm) using helium as the carrier gas. The following temperature program was used for the oven: 70 °C (initial, hold time 10 min) to 180 °C (final, hold time 20 min) at a rate of 5 °C min−1. The injector and the detector temperature were 220 and 230 °C, respectively. The gas flow rates for air, H2, and He were 300, 30, and 15 mL min−1, respectively. The split mode with a split ratio of 10:1 was used.
GC-MS analysis was performed using an instrument combined with an Agilent 5975C inert mass selective detector with a triple-Agilent GC 7890A axis detector. The same temperature program and gas flow rates used for the GC were used for the GC-MS. NMR spectra were recorded on a JOEL JNM-LA500 FT NMR at 500 MHz (1H) and at 125 MHz (13C) at room temperature, using deuterated chloroform (CDCl3) peak as an internal standard.
The E values were calculated according to the relationship:25
 | (1) |
Gene expression and purification of W110A TeSADH
W110A TeSADH was constructed and expressed in BL21(DE3) cells and purified as described previously.12a,21bPreparation of xerogel-encapsulated W110A TeSADH
Tetramethoxysilane (2035 μL, 1 mol eq.) water (470 μL, 2 mol eq.), and HCl (40 μL, 0.04 M) were vigorously shaken to form a sol. The enzyme stock containing NADP+ (1.0 mg), NADPH (1.0 mg) and W110A TeSADH (0.2–0.6 mg) in Tris–HCl buffer solution (400 μL, pH 8.0) was gently mixed with an equal volume of the sol for 5 seconds and left to stand for 24 h in a closed Eppendorf tube to allow gel aging. The hydrogel containing the immobilized enzyme and coenzymes was left to dry in open air for 24 h forming the xerogel-encapsulated W110A TeSADH.General procedure for racemization of enantiopure alcohol using xerogel-encapsulated W110 TeSADH
The enantiopure alcohol (0.05 mmol) and xerogel-encapsulated enzyme containing W110A TeSADH (0.2–0.6 mg), NADP+ (1.0 mg) and NADPH (1.0 mg) in a solvent (1.0 mL) were placed in a 1.5 mL Eppendorf tube. The reaction mixture was shaken at 200 rpm in a thermostat-controlled shaker at 50 °C for 24 h. The reaction mixture was extracted with diethyl ether (2 × 500 μL). The combined organic layers were dried over Na2SO4 and concentrated. For ee determination, the alcohols were derivatized by treatment with acetic anhydride (one drop) and pyridine (two drops) for 6 h prior to their analysis by chiral GC to determine the % conversion and % ee of the acetate ester derivatives.Procedures for racemization using free enzyme (aqueous and biphasic)
The enantiopure alcohol (0.05 mmol) W110A TeSADH (0.4 mg), NADP+ (1.0 mg) and NADPH (1.0 mg), were added to solvent (1.0 mL) in a round bottomed flask. The reaction in aqueous medium was conducted in Tris–HCl buffer (50 mM, pH 8.0)/acetonitrile (95:5 v/v) whereas the biphasic reaction in Tris–HCl buffer (50 mM, pH 8.0)/hexane (50:50 v/v). The reaction mixture was shaken at 200 rpm in a thermostat-controlled shaker at 50 °C for 24 h. The reaction mixture was extracted with diethyl ether (2 × 500 μL). The combined organic layers were dried over Na2SO4 and concentrated. For ee determination, the alcohols were derivatized by treatment with acetic anhydride (one drop) and pyridine (two drops) for 6 h prior to their analysis by chiral GC to determine the % conversion and % ee of the acetate ester derivatives.
General procedure for KR of racemic alcohols
Racemic alcohol [(rac)-1a–e, (0.05 mmol)], acyl donor (0.1 mmol), CALB (1 mg), and hexane (1.0 mL) were placed in a 1.5 mL Eppendorf tube. The mixture was shaken at 50 rpm at 50 °C for 3–6 h (monitored by GC equipped with a chiral column). A portion (50 μL) from the reaction was dried with Na2SO4 prior to analysis. Retention times of the resultant peaks were compared with standard samples of enantiopure alcohols and esters.General procedure for dual enzymatic DKR of racemic alcohols
Xerogel [containing W110A TeSADH (0.6 mg), NADPH (1.0 mg) and NADP+ (1.0 mg)], CALB (1.0 mg), (rac)-1a–e (0.03 mmol) and isopropenyl acetate (0.06 mmol) in hexane (1.0 mL) were placed in the same Eppendorf tube ensuring no physical contact between the two enzyme systems as shown in Fig. S2 of ESI.† The reaction mixture was shaken at 50 rpm at 50 °C for 24 h. The reaction content including the lipase and the xerogel were washed with ethyl acetate (2 × 500 μL). The combined organic layers were dried over Na2SO4 and concentrated. The ee of the ester derivatives was determined by chiral GC.Conflict of interest
The authors declare no competing financial interest.Acknowledgements
The authors acknowledge the support provided by King Abdulaziz City for Science and Technology (KACST) through the Science and Technology Unit at King Fahd University of Petroleum and Minerals (KFUPM), for funding this work through project No. 11-BIO1666-04, as part of the National Science, Technology, and Innovation Plan as well as baseline research funding offered to Prof. Hamdan through King Abdullah University of Science and Technology. The authors thank Prof. Claire Vieille, from the Department of Microbiology and Molecular Genetics at Michigan State University, for providing the plasmids of TeSADH.References
- (a) Y. K. Agrawal, H. G. Bhatt, H. G. Raval, P. M. Oza and P. J. Gorgoi, Mini-Rev. Med. Chem., 2007, 7, 451–460 CrossRef CAS PubMed; (b) G. C. Cotzias, M. H. Van Woert and L. M. Schiffer, N. Engl. J. Med., 1967, 276, 374–379 CrossRef CAS PubMed.
- (a) C. C. Gruber, I. Lavandera, K. Faber and W. Kroutil, Adv. Synth. Catal., 2006, 348, 1789–1805 CrossRef CAS; (b) J. H. Lee, K. Han, M.-J. Kim and J. Park, Eur. J. Org. Chem., 2010, 999–1015 CrossRef CAS; (c) M. Rachwalski, N. Vermue and F. P. J. T. Rutjes, Chem. Soc. Rev., 2013, 42, 9268–9282 RSC.
- (a) H. Pellissier, Tetrahedron, 2011, 67, 3769–3802 CrossRef CAS; (b) O. Verho and J.-E. Bäckvall, J. Am. Chem. Soc., 2015, 137, 3996–4009 CrossRef CAS PubMed.
- E. A. Díaz-Álvarez, L. Mesas-Sánchez and P. Dinér, Angew. Chem., Int. Ed., 2013, 52, 502–504 CrossRef PubMed.
- S. Y. Lee, J. M. Murphy, A. Ukai and G. C. Fu, J. Am. Chem. Soc., 2012, 134, 15149–15153 CrossRef CAS PubMed.
- J. Wang, D.-M. Do, G.-K. Chuah and S. Jaenicke, ChemCatChem, 2013, 5, 247–254 CrossRef CAS.
- O. A. Magnusson, M. Takwa, A. Hamberg and K. Hult, Angew. Chem., Int. Ed., 2005, 44, 4582–4585 CrossRef PubMed.
- (a) L. Borén, B. Martín-Matute, Y. Xu, A. Córdova and J.-E. Bäckvall, Chem.–Eur. J., 2006, 12, 225–232 CrossRef PubMed; (b) K. Engstrom, M. Vallin, P.-O. Syren, K. Hult and J.-E. Backvall, Org. Biomol. Chem., 2011, 9, 81–82 RSC; (c) M.-J. Kim, I. Y. Chung, K. Y. Choi, H. K. Lee, D. Kim and J. Park, J. Am. Chem. Soc., 2003, 125, 11494–11495 CrossRef CAS PubMed.
- M. M. Musa and R. S. Phillips, Catal. Sci. Technol., 2011, 1, 1311–1323 CAS.
- F. Hollmann, I. W. C. E. Arends and D. Holtmann, Green Chem., 2011, 13, 2285–2314 RSC.
- Y. Ni and J.-H. Xu, Biotechnol. Adv., 2012, 30, 1279–1288 CrossRef CAS PubMed.
- (a) M. M. Musa, R. S. Phillips, M. Laivenieks, C. Vieille, M. Takahashi and S. M. Hamdan, Org. Biomol. Chem., 2013, 11, 2911–2915 RSC; (b) C. C. Gruber, B. M. Nestl, J. Gross, P. Hildebrant, U. T. Bornscheuer, K. Faber and W. Kroutil, Chem.–Eur. J., 2007, 13, 8271–8276 CrossRef CAS PubMed.
- M. M. Musa, J. M. Patel, C. M. Nealon, C. S. Kim, R. S. Phillips and I. Karume, J. Mol. Catal. B: Enzym., 2015, 115, 155–159 CrossRef CAS.
- (a) D. Burdette and J. G. Zeikus, Biochem. J., 1994, 302, 163–170 CrossRef CAS PubMed; (b) D. Burdette, C. Vieille and J. G. Zeikus, Biochem. J., 1996, 316, 115–122 CrossRef CAS PubMed; (c) V. T. Pham and R. S. Phillips, J. Am. Chem. Soc., 1990, 112, 3629–3632 CrossRef CAS.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, Angew. Chem., Int. Ed., 2007, 46, 3091–3094 CrossRef CAS PubMed.
- V. Prelog, Pure Appl. Chem., 1964, 9, 119–130 CrossRef CAS.
- M. M. Musa, K. I. Ziegelmann-Fjeld, C. Vieille, J. G. Zeikus and R. S. Phillips, J. Org. Chem., 2007, 72, 30–34 CrossRef CAS PubMed.
- A. Ghanem and V. Schurig, Monatsh. Chem., 2003, 131, 1151–1157 CrossRef.
- J. Sangster, J. Phys. Chem. Ref. Data, 1989, 18, 1111–1226 CrossRef CAS.
- (a) L. Cao, L. V. Langen and R. A. Sheldon, Curr. Opin. Biotechnol., 2003, 14, 387–394 CrossRef CAS PubMed; (b) G. de Gonzalo, I. Lavandera, K. Faber and W. Kroutil, Org. Lett., 2007, 9, 2163–2166 CrossRef CAS PubMed; (c) S. Kara, D. Spickermann, A. Weckbecker, C. Leggewie, I. W. C. E. Arends and F. Hollmann, ChemCatChem, 2014, 6, 973–976 CrossRef CAS; (d) A. M. Klibanov, Curr. Opin. Biotechnol., 2003, 14, 427–431 CrossRef CAS PubMed; (e) M. V. Filho, T. Stillger, M. Müller, A. Liese and C. Wandrey, Angew. Chem., Int. Ed., 2003, 42, 2993–2996 CrossRef PubMed.
- (a) Unpublished results by Claire Vieille showed that TeSADH and TBADH are identical; (b) K. I. Ziegelmann-Fjeld, M. M. Musa, R. S. Phillips, J. G. Zeikus and C. Vieille, Protein Eng., Des. Sel., 2007, 20, 47–55 CrossRef CAS PubMed.
- M. Kitamura, M. Tokunaga and R. Noyori, J. Am. Chem. Soc., 1993, 115, 144–152 CrossRef CAS.
- A. Ghanem and H. Y. Aboul-Enein, Chirality, 2005, 17, 1–15 CrossRef CAS PubMed.
- A. L. Gemal and J. L. Luche, J. Am. Chem. Soc., 1981, 103, 5454–5459 CrossRef CAS.
- C. S. Chen, Y. Fujimoto, G. Girdaukas and C. J. Sih, J. Am. Chem. Soc., 1982, 104, 7294–7299 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Gas chromatograms, nuclear magnetic resonance spectra, and mass spectra are provided. See DOI: 10.1039/c6ra18895h |
| This journal is © The Royal Society of Chemistry 2016 |