
Insight into the acidic group-induced nitration mechanism of 2-methyl-4,6-dihydroxypyrimidine (MDP) with nitronium†
Kuan Wanga,
Jian-Gang Chen*a,
Bozhou Wangb,
Yueping Jib,
Fengyi Liua,
Zhao-Tie Liu*a,
Wenliang Wanga,
Zhong-Wen Liua,
Zhengping Haoac and
Jian Lu*b
aKey Laboratory of Applied Surface and Colloid Chemistry (MOE) and School of Chemistry & Chemical Engineering, Shaanxi Normal University, Xi'an, 710119, China. E-mail: jgchen@snnu.edu.cn; ztliu@snnu.edu.cn; Fax: +86-29-81530803; Fax: +86-29-81530802; Tel: +86-29-81530803 Tel: +86-29-81530802
bDepartment of Catalytic Technology, Institute of Xi'an Modern Chemistry, Xi'an, 710065, China. E-mail: lujian204@263.net; Tel: +86-29-88291213
cResearch Centre for Eco-Environmental Science, Chinese Academy of Sciences, Beijing, 100085, China
First published on 18th August 2016
Abstract
The strong desire for developing promising candidates of insensitive nitro energetic materials has spurred numerous attempts to discern the nitration details. The nitration mechanism of 2-methyl-4,6-dihydroxypyrimidine (MDP) with nitronium (NO2+) to form 2-dinitromethylene-5,5-dinitropyrimidine-4,6-dione is studied via DFT-B3LYP/6-311G(d,p) method. The possible nitration pathways are excavated and illustrated. Herein, a unique/incredible induction/enhancement of the co-existing acidic group of HSO4− to the targeted nitration is definitively proposed. The impact of the introduction order of four nitro groups (–NO2) on the title nitration is systematically demonstrated. It is suggested that the proposed induction of HSO4− may effectively promote/catalyze not only the NO2+ attack, but the H-transfer and H-abstraction as well, and thus dramatically decreases the activation free energy of the nitration system. Moreover, the NO2+ attack may be dynamically affected by the pre-introduced –NO2 and the resulted fluctuation of charge distribution in the pre-intermediates, which has been primarily supported via the calculated atomic charge, Coulomb attraction and Fukui function. It is indicated that the preferential dinitration on –CH3 with the induction of HSO4− (path A) is the most likely pathway. It is optimistically expected that the present study may provide a theoretical basis to the research and engineering tests of the title nitration, and promote the exploration of related insensitive energetic materials.
1. Introduction
Energetic materials are of especially great importance to economic and social development, and play an irreplaceable role in military affairs and national security. However, there exists a severe defect in the current extensively employed energetic materials: they are too sensitive to physical conditions (heat, impact and friction, etc.). Such a defect often leads to grievous casualties and severe accidents during production, transportation, storage and application, and has thus spurred numerous attempts to develop promising candidates for insensitive energetic materials to replace the traditional ones. The discovery of 1,1-diamino-2,2-dinitroethylene (FOX-7/DADNE) may be one of the most exciting events in the research of energetic materials.1 Owing to its superior performances of high energy density, high explosive and most importantly, lower sensitivity towards impact and friction, FOX-7 is increasingly accepted as a kind of new insensitive energetic materials. Therefore, in recent years much attention has been paid to investigating FOX-7 from various aspects, including the synthetic method,2–6 molecular/crystal structure,1,7–11 thermodynamic property,12–15 explosive performance,16,17 thermal decomposition18 and other performances/properties19–21 in recent years. However, so far quite few references can be found focusing on the synthesis mechanism of FOX-7. The absence of such significant details severely hinders the further research and application of FOX-7.Based on the currently available publications, it is found that the most popular strategy on the preparation of FOX-7 is started with the nitration of 2-methyl-4,6-dihydroxypyrimidine (MDP) in nitric–sulfuric acids, followed by the hydrolysis of the nitration product of 2-dinitromethylene-5,5-dinitropyrimidine-4,6-dione (N-MP).3–5 The typical preparation strategy of FOX-7 is shown in Scheme 1.
 | ||
| Scheme 1 Preparation strategy of FOX-7. | ||
Most nitration reactions to prepare energetic materials are found to correspond to electrophilic substitution mechanism, particularly in the nitration of the aromatics.22–26 Such mechanism was primarily verified by Esteves et al. via both simulation and experiment.23 Moreover, the nitration of benzene with NO2+ in the gas phase was found to follow a polar mechanism24 in the last stage of the electrophilic substitution, providing a further understanding to the nitration details. While in view that the actual nitration mostly occurred in nitric–sulfuric acids (in which the sulfuric acid may (partly) act as the solvent), the impacts of the reaction medium on the nitration, namely the solvent effect should be considered. Koleva et al.25 found that formamide may be used as a model solvent for the nitration in nitric–sulfuric acids because the dielectric constant of formamide (ε ≈ 108.94) is quite close to that of sulfuric acid (ε ≈ 100). A less polar medium of dimethylsulfoxide (ε = 46.8) was also employed as a model because of its similar structure to sulfuric acid. Interestingly, the obtained quite analogous potential energy surfaces in two model solvents indicated that the polarity of the medium may have slight/little impact on the nitration of benzene in mixed acid. Namely, the solvent effect in the nitration system may not be as noticeable as was expected. Such inference/assumption should be verified in the present work.
Since four nitro groups (–NO2) should be prerequisitly introduced to MDP during the formation of N-MP, the capture, separation and detection of the possible intermediates during the nitration process is hardly accomplished, resulting that the experimental investigation of the nitration details is considerably challenging. To the best of our knowledge, the detailed nitration mechanism of MDP has never been reported so far. As compared with the incompetency of experiential ways, the theoretical calculation/simulation, especially the DFT-B3LYP/6-311G(d,p) seems to be a versatile/promising method to the mechanism exploration of nitration systems from others27–29 and ours.30,31 Herein, the DFT-B3LYP/6-311G(d,p) method is employed aiming at the understanding of the nitration mechanism in the present work. While the following hydrolysis details of N-MP to form FOX-7 have also been investigated, and will be presented in another work.32
It is suggested that NO2+ may actually act as the nitration regent when the nitration reactions are performed in a concentrated nitric acid or mixed nitric–sulfuric acids.33 Moreover, we previously suggested that the co-existed acidic group of HSO4− may promote the nitration of triazol-3-one (TO) with NO2+.30 It is eagerly expected and optimistically supposed that such promotion/enhancement may also exist in the nitration of MDP with NO2+, which needs to be verified.
In the present work, the HSO4− induced nitration mechanism of MDP with NO2+ to form N-MP is systematically investigated via DFT-B3LYP/6-311G(d,p). The possible nitration pathways are respectively excavated, in which the hypothesized induction/enhancement of HSO4− to the titled nitration is proposed and theoretically verified. Moreover, the impact of –NO2 attacking order on the nitration is also discerned, through which the preferential nitration pathway was illustrated. The present paper is expected to provide a better understanding on the nitration details of MDP, and will contribute great to the research and engineering tests of the preparation of FOX-7 and other nitro energetic materials.
2. Computational details
The calculations were performed using the Gaussian 09 software package.34 The DFT-B3LYP/6-311G(d,p) method was employed to optimize the geometries of the reactant complexes, intermediates, transition states and products in the nitration system.35,36 B3LYP/6-311G(d,p) was also applied to analyze the vibrational frequency, via which whether the obtained structures are the ones of the transition states or local minima points is verified, and the zero-point vibrational energy (ZPE) was predicted. Intrinsic reaction coordinate (IRC)37 analysis was carried out to ensure that the structure of every transition state obtained does connect the corresponding reactant and product of the (elementary) reaction. Moreover, B3LYP-D3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 38,39 with a larger 6-311++G(3df,3pd) basis set was intentionally used to calculate the single-point energies of all the species to obtain more reliable energy results based on the optimized geometries. The Mulliken atomic charges40 of MDP and the relevant intermediates were also calculated via B3LYP/6-311G(d,p). The relative free energies of different stationary points obtained via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p) are listed in Table 1. Additionally, the solvent effect was also simulated and primarily evaluated using the conductor-like polarizable continuum model (CPCM).41
38,39 with a larger 6-311++G(3df,3pd) basis set was intentionally used to calculate the single-point energies of all the species to obtain more reliable energy results based on the optimized geometries. The Mulliken atomic charges40 of MDP and the relevant intermediates were also calculated via B3LYP/6-311G(d,p). The relative free energies of different stationary points obtained via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p) are listed in Table 1. Additionally, the solvent effect was also simulated and primarily evaluated using the conductor-like polarizable continuum model (CPCM).41
| Path A | Path B | Path C | ||||||
|---|---|---|---|---|---|---|---|---|
| No. | Species | ΔG | No. | Species | ΔG | No. | Species | ΔG |
| a ZPG was obtained via DFT at the B3LYP/6-311G(d,p) level. The energy value (ΔG) was obtained at the B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p) level.b Here, HSO4− is abbreviated or denoted as S.c HNO3 is denoted as N.d H2SO4 is denoted as SH. | ||||||||
| A-1 | MDP + 4Sb + 4Nc | 0.0 | B-1 | MDP + 4S + 4N | 0.0 | C-1 | MDP + 4S + 4N | 0.0 |
| A-2 | A-IM1 + 3S + 4N | −8.5 | B-2 | B-IM1 + 3S + 4N | −1.5 | C-2 | Ca-IM1 + 2S + 3N + SH | −180.7 |
| A-3 | A-TS1 + 3S + 4N | 5.3 | B-3 | B-TS1 + 3S + 4N | 0.3 | C-3 | Ca-TS1 + 2S + 3N + SH | −168.2 |
| A-4 | A-IM2 + 3S + 4N | −10.8 | B-4 | B-IM2 + 3S + 4N | −15.0 | C-4 | Ca-IM2 + 2S + 3N + SH | −183.0 |
| A-5 | A-IM3 + 3S + 3N | −130.3 | B-5 | B-IM3 + 3S + 4N | −14.7 | C-5 | Ca-IM3 + 2S + 3N + SH | −183.4 |
| A-6 | A-TS2 + 3S + 3N | −124.7 | B-6 | B-TS2 + 3S + 4N | −10.9 | C-6 | Ca-TS2 + 2S + 3N + SH | −180.6 |
| A-7 | A-IM4 + 3S + 3N | −169.4 | B-7 | B-IM4 + 3S + 4N | −11.2 | C-7 | Ca-IM4 + 2S + 3N + SH | −181.4 |
| A-8 | A-IM5 + 2S + 3N + SHd | −180.9 | B-8 | B-IM5 + 3S + 3N + SH | −141.1 | C-8 | Ca-IM5 + 2S + 2N + 2SH | −303.7 |
| A-9 | A-TS3 + 2S + 3N + SH | −178.0 | B-9 | B-TS3 + 3S + 3N + SH | −112.0 | C-9 | Ca-TS3 + 2S + 2N + 2SH | −274.9 |
| A-10 | A-IM6 + 2S + 3N + SH | −194.3 | B-10 | B-IM6 + 3S + 3N + SH | −159.3 | C-10 | Ca-IM6 + 2S + 2N + 2SH | −322.5 |
| A-11 | A-IM7 + 3S + 2N + SH | −205.1 | B-11 | B-IM7 + 2S + 3N + SH | −184.0 | C-11 | Ca1-IM7 + S + 2N + 2SH | −350.0 |
| A-12 | A-TS4 + 3S + 2N + SH | −182.5 | B-12 | B-TS4 + 2S + 3N + SH | −168.5 | C-12 | Ca1-TS4 + S + 2N + 2SH | −337.0 |
| A-13 | A-IM8 + 3S + 2N + SH | −208.2 | B-13 | B-IM8 + 2S + 3N + SH | −173.9 | C-13 | Ca1-IM8 + S + 2N + 2SH | −348.3 |
| A-14 | A-IM9 + 2S + 2N + SH | −318.7 | B-14 | B-IM9 + 2S + 2N + 2SH | −292.3 | C-14 | Ca1-IM9 + S + N + 3SH | −454.9 |
| A-15 | A-TS5 + 2S + 2N + SH | −314.7 | B-15 | B-TS5 + 2S + 2N + 2SH | −271.6 | C-15 | Ca1-TS5 + S + N + 3SH | −434.6 |
| A-16 | A-IM10 + 2S + 2N + SH | −334.3 | B-16 | B-IM10 + 2S + 2N + 2SH | −312.6 | C-16 | Ca1-IM10 + S + N + 3SH | −473.6 |
| A-17 | A-IM11 + S + 2N + 2SH | −346.2 | B-17 | B-IM11 + S + 2N + 2SH | −333.0 | C-17 | Ca2-IM7 + S + 2N + 2SH | −346.0 |
| A-18 | A-TS6 + S + 2N + 2SH | −344.7 | B-18 | B-TS6 + S + 2N + 2SH | −318.2 | C-18 | Ca2-TS4 + S + 2N + 2SH | −343.5 |
| A-19 | A-IM12 + S + 2N + 2SH | −355.8 | B-19 | B-IM12 + S + 2N + 2SH | −331.8 | C-19 | Ca2-IM8 + S + 2N + 2SH | −357.2 |
| A-20 | A-IM13 + S + N + 3SH | −461.1 | B-20 | B-IM13 + S + N + 2SH | −445.3 | C-20 | Ca2-IM9 + 2S + N + 2SH | −356.4 |
| A-21 | A-TS7 + S + N + 3SH | −439.2 | B-21 | B-TS7 + S + N + 2SH | −433.5 | C-21 | Ca2-TS5 + 2S + N + 2SH | −333.9 |
| A-22 | A-IM14 + S + N + 3SH | −487.3 | B-22 | B-IM14 + S + N + 2SH | −475.4 | C-22 | Ca2-IM10 + 2S + N + 2SH | −357.9 |
| A-23 | A-IM15 + N + 3SH | −512.9 | B-23 | B-IM15 + S + N + 2SH | −497.6 | C-23 | Ca2-IM11 + S + N + 2SH | −474.2 |
| A-24 | A-TS8 + N + 3SH | −509.7 | B-24 | B-TS8 + S + N + 2SH | −495.3 | C-24 | Ca2-TS6 + S + N + 2SH | −468.4 |
| A-25 | A-IM16 + N + 3SH | −526.3 | B-25 | B-IM16 + S + N + 2SH | −511.3 | C-25 | Ca2-IM12 + S + N + 2SH | −490.2 |
| A-26 | A-IM17 + 4SH | −620.3 | B-26 | B-IM17 + S + 3SH | −500.7 | C-26 | Cb-IM1 + 2S + 3N + SH | −176.3 |
| A-27 | A-TS9 + 4SH | −600.7 | B-27 | B-TS9 + S + 3SH | −481.1 | C-27 | Cb-TS1 + 2S + 3N + SH | −161.5 |
| A-28 | A-IM18(N-MP) + 4SH | −637.0 | B-28 | B-IM18 + S + 3SH | −502.7 | C-28 | Cb-IM2 + 2S + 3N + SH | −176.4 |
| B-29 | B-IM19 + 3SH | −624.7 | C-29 | Cb-IM3 + 2S + 2N + SH | −290.5 | |||
| B-30 | B-TS10 + 3SH | −615.5 | C-30 | Cb-TS2 + 2S + 2N + SH | −282.6 | |||
| B-31 | B-IM20 + 3SH | −639.8 | C-31 | Cb-IM4 + 2S + 2N + SH | −324.3 | |||
| B-32 | N-MP + 4SH | −637.0 | ||||||
3. Results and discussion
The formation of NO2+ in the concentrated nitric acid or nitric–sulfuric acids (HNO3 + 2H2SO4 → H3O+ + NO2+ + 2HSO4−) has been investigated for years.33,42 It was suggested that NO2+ may actually act as the nitration reagent since NO2+ is found to be a more efficient electrophile than HNO3. Moreover, the HSO4− was found to play an important role since it may effectively promote/catalyze the nitration in our recent works.30,31 Therefore, the nitration of MDP in nitric–sulfuric acids may thus be simplified as that of MDP with NO2+ in the presence of HSO4−. Additionally, inspired by the publication from Koleva et al.25 it is supposed that the solvent effect from mixed acid to the titled nitration may be inapparent. Therefore, the nitration in gas phase is mainly discussed in the present work. The solvent effect is also investigated so as to explore and understand the impacts of the medium on the titled nitration. Based on such cognition, the nitration mechanism of MDP with NO2+ to form N-MP is investigated, so does the impact of the nitration order. In view of the Mulliken atomic charges of MDP (Table S1†) and our recent work,30 we focus our interests on the C-nitration pathway(s), namely only the attacking of methylene C4 and methyl C10 in MDP by NO2+ are considered. Such C-nitration pathways are systematically discussed in the present work.3.1 Nitration mechanism of MDP
We suggest that the nitration of MDP with NO2+ may be induced/catalyzed by the co-existed HSO4−. Such HSO4− induced nitration of MDP is denoted as path A. The potential energy profiles and the corresponding optimized geometries are shown in Fig. 1 and 2, respectively. While to verify the expected induction/enhancement of HSO4−, a hypothetical direct nitration path way of MDP (without any assistance/cooperation of HSO4− in the NO2+ attacking step, denoted as path A1′) is also studied and presented in Fig. S1.†
 | ||
| Fig. 1 Schematic energy diagram for potential energy surface of the preferential dinitration on methyl group of MDP in the gas phase predicted via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p). For brevity, the systems with different species are numbered/denoted from “A-1” to “A-28”. The correspondence of the serial number and the systems is listed in Table 1. Insert (a): the potential energy surfaces of the first NO2+ attacking step of MDP. Insert (b): the potential energy surfaces of the H-transferring step of MDP. | ||
 | ||
| Fig. 2 Optimized geometries of species in the preferential dinitration on methyl group of MDP in the gas phase calculated via B3LYP/6-311G(d,p) (bond lengths are in angstrom). The serial number (in parentheses) represents the system of corresponding species listed in Table 1. | ||
As shown in Fig. S1,† it is seen that the NO2+ attacking and the hydrogen transferring (H-transferring) may occur simultaneously in the direct nitration of MDP (path A′). The activation free energy (ΔG≠) associated with this NO2+ attacking is 62.6 kcal mol−1, indicating that path A1′ is severely unlikely to occur. While as shown in Fig. 1 and 2, it is surprisingly found that the ΔG≠ for the NO2+ attacking (A-IM3 → A-IM4) is merely 5.6 kcal mol−1. An unexpected sharp decrease of 57 kcal mol−1 is achieved, indicating that the nitration may be effectively promoted/enhanced by HSO4−. Such NO2+ attacking is definitely much more favorable than that in path A1′ (Fig. S1†).
The schematic energy diagram of the HSO4−-induced NO2+ attacking to MDP is shown in Fig. S2.† As shown in Fig. S2,† the ΔG≠ for the HSO4−-induced NO2+ attacking is 46.1 kcal mol−1, leading to a decrease of 16.5 kcal mol−1 as compared with that in path A1′. Such a high ΔG≠ value indicates that path A1 (Fig. S2†) is unlikely to occur. It is supposed that the decrease in ΔG≠ may be derived from the induction of HSO4−. However, such decrease is not as much as what was expected.
We supposed that if H11 in C10 transfers to N5, the HSO4−-induced nitration step may occur more likely for that such H-transferring may obviously decrease the steric hindrance and increase the nucleophilicity of C10. The schematic energy diagram of the direct H-transferring (without the assistance of HSO4−) is shown in Fig. S3† (ΔG≠ = 53.8 kcal mol−1). While as compared with the HSO4−-induced H-transferring (A-IM1 → A-IM2, Fig. 2, ΔG≠ = 13.8 kcal mol−1), a dramatical decrease of approximately 40.0 kcal mol−1 is obtained (insert (b), Fig. 1), indicating that the HSO4−-induced H-transferring is much more likely to occur. In view that similar induction/promotion of HSO4− is also found in the H-transferring of the mono-nitrated MDP (A-IM5 → A-IM6, Fig. 2) if compared with the direct H-transferring of the mono-nitrated MDP (A3′-IM1 → A3′-IM2, Fig. S4†), we believe that the promotion/enhancement of the HSO4− induction to H-transferring is verified.
We supposed that the next NO2+ attacking and the H-abstracting may occur simultaneously after A-IM2 is obtained. The HSO4−-induced NO2+ attacking along with the H-abstracting is shown in Fig. 2 (A-IM3 → A-IM4). As shown in Fig. 2, it is seen that with the approach of the HSO4−⋯NO2+ to C10 and H21, an eight-membered transition state of A-TS2 is formed by C10–N13–O23–S15–O22–H21–N5–C2, in which one NO2+ (N13) is added (to C10), and one hydrogen (H21) is synchronously transferred (from N5 to O22).
The NO2+ attacking step in every nitration path is compared and discussed. The ΔG≠ in A-IM3 → A-IM4 (Fig. 2) is much lower than that in either A1′-IM1 → A1′-IM2 (Fig. S1†) or A1-IM1 → A1-IM2 (Fig. S2†). Clearly (insert (a), Fig. 1), A-IM3 → A-IM4 is much more favorable than A1′-IM1 → A1′-IM2 and A1-IM1 → A1-IM2. It is inferred that the H-transferring must be the prerequisite to the nitration of MDP. More importantly, both the H-transferring and the NO2+ attacking must be synergistically promoted/enhanced by HSO4− via a unique induction. Such hypothesis is well supported by the results of the Mulliken atomic charges of the relevant pre-intermediates in the NO2+ attacking steps listed in Table S1.† In Table S1,† the atomic charge of C10 in A-IM3 is the most negative among that in the three pre-intermediates. It is indicated that the C10 in A-IM3 may have the best nucleophilicity to be favorably attacked by NO2+. As compared with the direct nitration (Fig. S1†), it is seen that without H-transferring of MDP (C10 to N5 in Fig. S2†), the nucleophilicity of C10 may also distinctly increase if the induction of HSO4− to the NO2+ attacking is considered. While the increase of the atomic charge of C10 in A1-IM1 is found much inferior to that in A-IM3, in which the H-transferring and HSO4− induced NO2+ attacking are both considered.
After the mono-nitration of MDP in Fig. 2 (A-IM3 → A-IM4), and the subsequent HSO4− induced H-transferring in mono-nitrated MDP (A-IM5 → A-IM6), the second NO2+ attacking occurs (A-IM7 → A-IM8). While it seems that HSO4− does not participate the second NO2+ attacking partly due to the steric hindrance from the prior-introduced –NO2 on the same C atom of C10. Luckily, the ΔG≠ in the second NO2+ attacking step is found fairly moderate (22.6 kcal mol−1). Possible reason is that the second NO2+ may be attracted by the O atom in the previously introduced –NO2, resulting in the formation of the complex (A-IM7, Fig. 2) and facilitating the subsequent attacking of NO2+ to C10 site (A-IM7 → A-IM8, Fig. 2). When the rest H atom on C10 is abstracted, the preferential dinitration on this methyl group is achieved.
It is reported that when NO2+ is employed as the nitration agent, the nitro group in the product may be further protonated to –NOOH, followed by that the proton in the –NOOH may be easily abstracted in the presence of acidic group(s).24,30,31 It is inferred that when the rest H atom on C10 is transferred to the nitro group, the H-abstracting may be successfully performed. Such process is studied in the present work. The schematic energy diagram of the H-transferring of the dinitrated MDP is shown in Fig. S5† (from C10 to O20). It is seen that without the assistance of HSO4−, the ΔG≠ in the H-transferring step is found to be 40.3 kcal mol−1 (A4′-IM1 → A4′-IM2, Fig. S5†). While when the HSO4− induction/promotion is considered, it is clearly seen that ΔG≠ in the H-abstracting step (A-IM9 → A-IM10, Fig. 1 and 2) dramatically decreases to 2.3 kcal mol−1. Definitely, the HSO4− induced H-abstracting in Fig. 2 (A-IM9 → A-IM10) is much more favorable than the H-transferring in Fig. S5† (A4′-IM1 → A4′-IM2). We thus strongly suggest that the H-abstracting may be effectively induced/promoted by HSO4−, which is readily adopted in the rest steps of path A.
The successful obtaining of A-IM10 indicates that C10 in MDP has been preferentially dinitrated. Hereafter, the nitration on C4 is discussed.
We once supposed that the next nitration step may be similar to A-IM3 → A-IM4, in which the NO2+ attacking on the C4 may occur simultaneously with the H-abstracting in such step. However, such assumption was not supported by the calculation, though several methods were attempted and weeks of time were paid. While, it can be inferred that the H atom(s) on C4 may be distinctly activated by the two adjacent carbonyl groups, for that the H-abstracting on the C4 is found quite likely to occur in the presence of HSO4− (A-IM11 → A-IM12, ΔG≠ = 1.5 kcal mol−1, Fig. 2 and Table 1). Subsequently, the NO2+ attacking to C4 site occurs (A-IM13 → A-IM14), in which the ΔG≠ is 21.9 kcal mol−1. In a word, as to the introduction of the third –NO2 to the methylene C4 in MDP (A-IM11 → A-IM14) is concerned, though the H-abstracting and the NO2+ attacking may not occur simultaneously, it is suggested that A-IM11 → A-IM14 is definitely promoted by HSO4−.
The introduction of the fourth –NO2 to C4 (A-IM15 → A-IM18) is found to be quite similar to that of the third one. The fourth nitration is supposed to begin with the HSO4−-induced H-abstracting step (A-IM15 → A-IM16) with a ΔG≠ of 3.2 kcal mol−1. Followed by the NO2+ attacking to C4 site (A-IM17 → A-IM18), which has a ΔG≠ of 19.6 kcal mol−1, after which the targeted nitration product of N-MP is eventually achieved. Clearly, the second NO2+ attacking to C4 step (path A, A-IM7 → A-IM8) is the rate-determining step, as shown in Fig. 1.
Based on the above mentioned calculation, it is strongly suggested that the co-existed acidic group of HSO4− may effectively induce/catalyze the nitration process. We find that besides the NO2+ attacking step is promoted as was expected, the H-transferring as well as the H-abstracting is surprisingly enhanced by HSO4− in the nitration process in path A. Moreover, it is reported that FOX-7 could been prepared via the nitration of MDP in the nitric–sulfuric acids with high yield, even though the reaction time is relative short along with the temperature is low.5,6 Such facts indicated that the activity free energy of the nitration should be considerably low, and the HSO4−-induced path A may be more favorable way than the direct path, which is in very good agreement with the experimental results.6 Additionally, in view of the fact that a higher H2SO4 concentration may benefit an easier formation of NO2+ along with a more difficult dissociation of H2SO4 to form HSO4−, the concentration of H2SO4 should be well controlled.
To the best of our knowledge, similar acidic group-induced nitration has not been reported by others so far. It is such unique induction/enhancement of HSO4− to the NO2+ attacking, H-transferring and H-abstracting that dramatically decreases the ΔG≠, effectively promotes the formation of N-MP, and thus is specifically preferred in the rest part of the present work.
 | ||
| Fig. 3 Optimized geometries of species in the preferential dinitration on methylene group of MDP calculated via B3LYP/6-311G(d,p) (bond lengths are in angstrom). The serial number (in parentheses) represents the system of corresponding species listed in Table 1. | ||
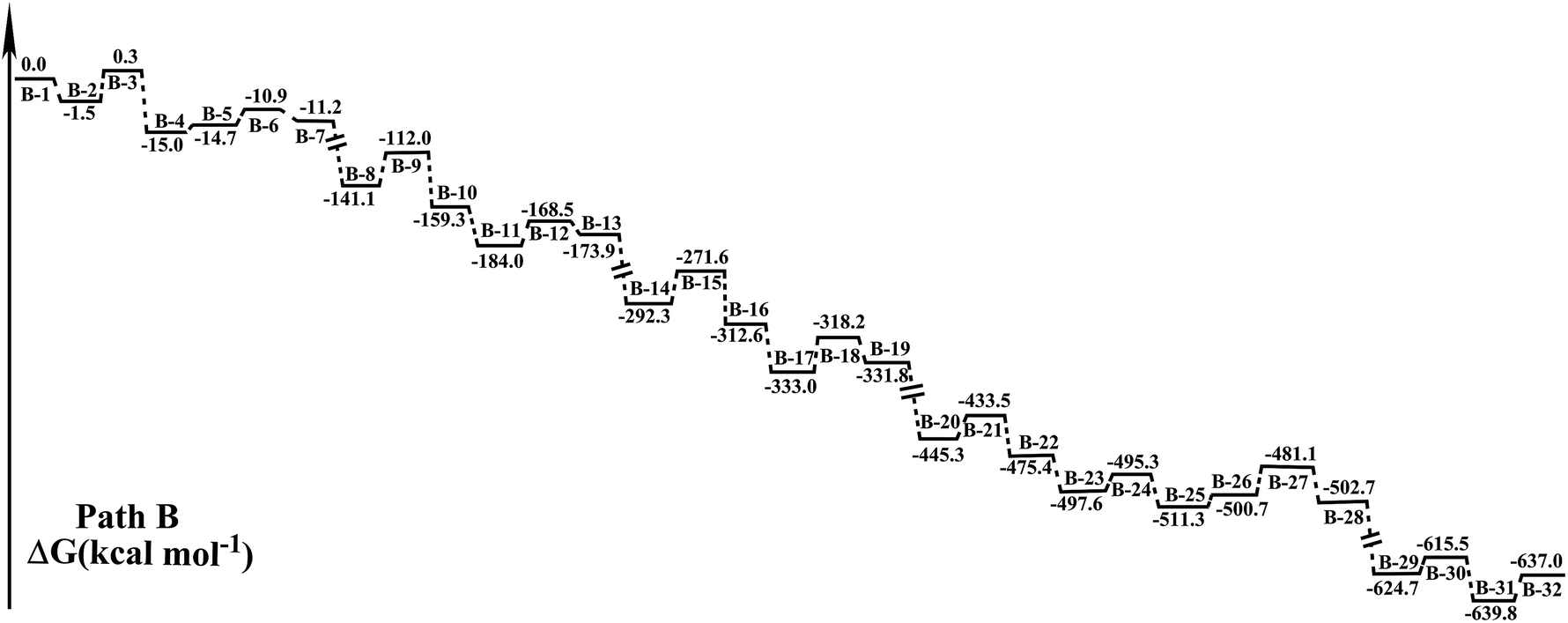 | ||
| Fig. 4 Schematic energy diagram for potential energy surface of the preferential dinitration on methylene group of MDP in the gas phase predicted via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p). The systems with different species are numbered/denoted from “B-1” to “B-32”. The correspondence of the serial number and the systems is listed in Table 1. | ||
As is seen in Fig. 3 and 4, the introduction of the two –NO2 to C4 (B-IM1 → B-IM10) in this path is exactly similar to that in path A (A-IM11 → A-IM18) except that the H-abstracting (B-IM1 → B-IM4) may be aided by a cooperated H-transferring. Clearly, path B starts with an H-transferring-aided H-abstracting step, which is somewhat different to what is mentioned in path A. During the approaching of HSO4− to MDP, the close of H13 on C4 to O18 occurs simultaneously with that of H19 to O8. After an eight-centered transition state of B-TS1, B-IM2 forms with a ΔG≠ of 1.8 kcal mol−1. Then H19 on O8 is easily abstracted by O18 in HSO4− (B-IM3 → B-IM4, ΔG≠ = 3.8 kcal mol−1) and the H-abstracting is achieved, followed by C4 is attacked. It is found that the ΔG≠ in the subsequent first NO2+ attacking to C4 (B-IM5 → B-IM6) is 29.1 kcal mol−1, which is higher than that in A-IM13 → A-IM14 in path A (ΔG≠ = 20.9 kcal mol−1). After the HSO4−-induced H-abstracting (B-IM7 → B-IM8, ΔG≠ = 15.5 kcal mol−1) and the second NO2+ attacking to C4 (B-IM9 → B-IM10, ΔG≠ = 20.7 kcal mol−1), the methylene C4 in MDP is preferentially dinitrated.
The introduction of the two –NO2 to C10 in this path (B-IM11 → B-IM20) is similar to that in path A (A-IM1 → A-IM10, Fig. 1), and mainly includes five steps. It is seen that the ΔG≠ in the HSO4−-induced H-transferring (B-IM11 → B-IM12) is 14.8 kcal mol−1. After the H-transferring is performed, the NO2+ attacking to C10 occurs simultaneously with the H-abstracting (B-IM13 → B-IM14, ΔG≠ = 11.8 kcal mol−1), which leads to the introduction of the third –NO2 to MDP. After the HSO4−-induced H-transferring (B-IM15 → B-IM16, ΔG≠ = 2.3 kcal mol−1), C10 is attacked by NO2+ (B-IM17 → B-IM18, ΔG≠ = 19.6 kcal mol−1). Followed by H13 in C10 is abstracted by HSO4− (B-IM19 → B-IM20, ΔG≠ = 9.2 kcal mol−1), N-MP forms.
 | ||
| Fig. 5 Optimized geometries of species in the crossed nitration on methyl and methylene group of MDP calculated via B3LYP/6-311G(d,p) (bond lengths are in angstrom). The serial number (in parentheses) represents the system of corresponding species listed in Table 1. | ||
 | ||
| Fig. 6 Schematic energy diagram for potential energy surface of the crossed nitration on methyl and methylene group of MDP predicted via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p). The systems with different species are numbered/denoted from “C-1” to “C-25”. The correspondence of the serial number and the systems is listed in Table 1. | ||
Path Ca. The introduction of the first –NO2 to C10 in this path is identical to that in path A (A-IM1 → A-IM4, Fig. 2), in which the mononitrated MDP is obtained, followed by Ca-IM1 (Fig. 5) forms. Then, the second –NO2 may be introduced to C4, resulting in the formation of Ca-IM6 (Fig. 2). While since the third –NO2 may be introduced to either C4 or C10, the corresponding paths are denoted as Ca1 and Ca2. Clearly, the introduction of the third –NO2 may dominate how the last –NO2 is introduced. If the former follows Ca1 path, the introduction of the last –NO2 may be identical to that in path B (B-IM15 → B-IM20). By the same token, if the former follows Ca2 path, the last –NO2 may be introduced identically to that in path A (A-IM15 → A-IM18).
As shown in Fig. 5 and 6, after H-transferring (Ca-IM1 → Ca-IM2, ΔG≠ = 12.5 kcal mol−1) aided H-abstracting process (Ca-IM3 → Ca-IM4, ΔG≠ = 2.8 kcal mol−1), H13 in C4 is abstracted, and Ca-IM4 is obtained. Next, C4 in Ca-IM5 is attacked by NO2+ (Ca-IM5 → Ca-IM6, ΔG≠ = 28.8 kcal mol−1), after which the second –NO2 is introduced. Subsequently, the introduction of the third –NO2 may either follow path Ca1, which includes two successive steps as the H-abstracting (Ca1-IM7 → Ca1-IM8, ΔG≠ = 13.0 kcal mol−1) and NO2+ attacking (Ca1-IM9 → Ca1-IM10, ΔG≠ = 20.3 kcal mol−1), or follow path Ca2, which includes three successive steps as the H-transferring (Ca2-IM7 → Ca2-IM8, ΔG≠ = 2.5 kcal mol−1), NO2+ attacking (Ca2-IM9 → Ca2-IM10, ΔG≠ = 22.5 kcal mol−1), and H-abstracting (Ca2-IM11 → Ca2-IM12, ΔG≠ = 5.8 kcal mol−1). Finally, the introduction of the last –NO2 may either follow path Ca1, which is identical to that in path B (B-IM15 → B-IM20), or follow path Ca2, which is identical to that in path A (A-IM15 → A-IM18).
Path Cb. The introduction of the first –NO2 to C4 in this path is identical to that in path B (B-IM1 → B-IM6, Fig. 3), followed by Cb-IM1 (Fig. 5) forms. Next, the second –NO2 may be introduced to C10, resulting in the formation of Cb-IM4. Since Cb-IM4 is exactly the complex of Ca-IM6 with H2SO4, the subsequent nitration process in path Cb is actually identical to that in path Ca. In view of this, the pathway similar to path Ca1 in the introduction of the last two –NO2 is denoted as path Cb1, and the pathway similar to path Ca2 is denoted as path Cb2.
In this way the possible pathways (paths A to C) in the C-nitration of MDP with NO2+ to form N-MP have been systematically demonstrated. Since four nitro groups are prerequisitly needed to be introduced during the formation of N-MP, the impacts of the introduction order of nitro groups on nitration of MDP should be discerned.
 | ||
| Fig. 7 The activation free energy (ΔG≠) in the four nitronium attacking steps (① to ④) in path A, path B and (the four sub-pathways of) path C in the gas phase predicted via B3LYP-D3/6-311++G(3df,3pd)//B3LYP/6-311G(d,p). The insert shows the ΔG≠ in the first nitronium attacking in the paths of preferential dinitration on methyl group with/without the induction of HSO4−. | ||
As shown in Fig. 7, firstly, it is clear that path A must be more likely to occur, indicating that the C10 in MDP may be preferentially dinitrated. Secondly, it is found that the step ③ in every path may be the rate-determining step during the nitration of MDP. Thirdly, the ΔG≠ of step ② is found to be very close in paths A to C, so does step ④, indicating that the steps ② and ④ may be little affected by the nitration order. Additionally, the ΔG≠ of step ① is found to increase in step with the increase of the number of nitro group on C4 in MDP. It is suggested that the NO2+ attacking may be dynamically affected by the introduction of –NO2 and the resulted fluctuation of the charge distribution in the corresponding pre-intermediates.
To discern the reason why path A is the most favorable way among the possible paths, the rate-determining step ③ in paths A to C is further investigated, and the atomic charge of every corresponding pre-intermediate is calculated via B3LYP/6-311G(d,p). The calculated atomic charges (Mulliken,40 NPA,43,44 CHELPG45 and Hirshfeld46) of the pre-intermediates in the step ③ in paths A to C are shown in Tables 2 and S2,† in which the distance between the pre-reactive atoms in every path is also listed.
| Methods | Species | Q/ea | r/mb | F/Nc | ||||
|---|---|---|---|---|---|---|---|---|
| C4 | N16 | O8 | C4–N16 | O8–N16 | C4–N16 | O8–N16 | ||
| a Q, the atomic charge of the pre-intermediates in the step ③ in paths A to C for the nitration of MDP with NO2+ calculated via B3LYP/6-311G(d,p). To calculate the Coulomb force, the charge unit of e should be transformed to Coulomb (C) with a relationship as: 1e = 1.602 × 10−19 C.b r, the distance between atoms C4 and N16 or atoms O8 and N16 is calculated via B3LYP/6-311G(d,p).c F, the Coulomb attraction between atoms C4 and N16 or atoms O8 and N16 is calculated by Coulomb's law (F = −kQ1Q2/r2), where the constant k is 9.0 × 109 N m2 C−2 if other variables take their standard international units. | ||||||||
| Mulliken | A-IM13 | −0.214 | 0.441 | −0.320 | 3.325 × 10−10 | 1.594 × 10−10 | 1.972 × 10−10 | 1.283 × 10−9 |
| B-IM15 | −0.207 | 0.435 | −0.290 | 3.477 × 10−10 | 1.498 × 10−10 | 1.721 × 10−10 | 1.299 × 10−9 | |
| Ca-IM5 | −0.202 | 0.436 | −0.291 | 3.475 × 10−10 | 1.507 × 10−10 | 1.685 × 10−10 | 1.291 × 10−9 | |
| NPA | A-IM13 | −0.326 | 0.732 | −0.482 | 3.325 × 10−10 | 1.594 × 10−10 | 4.986 × 10−10 | 3.207 × 10−8 |
| B-IM15 | −0.348 | 0.749 | −0.436 | 3.477 × 10−10 | 1.498 × 10−10 | 4.971 × 10−10 | 3.361 × 10−8 | |
| Ca-IM5 | −0.331 | 0.748 | −0.438 | 3.475 × 10−10 | 1.507 × 10−10 | 4.736 × 10−10 | 3.332 × 10−8 | |
| CHELPG | A-IM13 | −0.574 | 0.850 | −0.483 | 3.325 × 10−10 | 1.594 × 10−10 | 1.019 × 10−9 | 3.732 × 10−8 |
| B-IM15 | −0.617 | 0.861 | −0.445 | 3.477 × 10−10 | 1.498 × 10−10 | 1.014 × 10−9 | 3.944 × 10−8 | |
| Ca-IM5 | −0.607 | 0.846 | −0.436 | 3.475 × 10−10 | 1.507 × 10−10 | 9.822 × 10−10 | 3.751 × 10−8 | |
We supposed that a stronger attraction between C4 and N16, along with a weaker attraction between O8 and N16, may contribute to an easier attacking of NO2+ to C4, and a lower ΔG≠ in the step ③. Such attraction is believed to be dependent on both the quantity of electricity (Q) and the distance (r) between the two atoms. As is clearly seen in Table 2, for the Mulliken atomic charge (the first three rows), C4 in A-IM13 (path A) is more negative than that of either B-IM5 or Ca-IM5, and N16 in the former is more positive than that in the latter two. Simultaneously, the distance between atoms C4 and N16 in A-IM13 is the shortest among the pre-intermediates. Thus it is inferred that the attraction between atoms C4 and N16 in A-IM13 may be stronger than that in B-IM5 or Ca-IM5. However, it is difficult to directly and qualitatively compare/evaluate the attraction between atoms O8 and N16 among the three pre-intermediates. Since though the distance between atoms O8 and N16 in A-IM13 is the longest (which may contribute to a weaker attraction), the Q in the two atoms in A-IM13 is the biggest among the three pre-intermediates (which may promote a stronger attraction). Thus the impacts of both the Q and r should be synergistically considered. Here, the Coulomb's law is intentionally employed, through which the attraction between atoms (C4 and N16 versus O8 and N16) is evaluated.
The Coulomb attraction was estimated based on the atomic charge obtained via the Mulliken method. The results are listed in Table 2. As is clearly seen in Table 2, among the three pre-intermediates listed, the Coulomb attraction between atoms C4 and N16 in A-IM13 is the strongest. Meanwhile, the Coulomb attraction between atoms O8 and N16 in A-IM13 is the weakest (as what is expected). Such two factors may contribute to an easier attacking of NO2+ to C4 in A-IM13, and result in a lower ΔG≠ in the rate-determining step ③. For example, the ΔG≠ in step ③ in path A is 20.9 kcal mol−1, which is approximately 8 kcal mol−1 lower than that in paths B and C.
The Coulomb attraction was also calculated using the atomic charge from the NPA and CHELPG methods (shown in Table 2). As is seen in Table 2, the Coulomb attraction derived from either NPA or CHELPG method is the strongest between atoms C4 and N16 in A-IM13, and is the weakest between atoms O8 and N16 in A-IM13. Fortunately, such results are in very good agreement with what were obtained from the Mulliken method, indicating that the path A is more likely to occur.
However, as is seen in Table S2,† the Coulomb attraction between atoms C4 and N16 in A-IM13 obtained via Hirshfeld method is not the strongest. Such result is not in accordance with what obtained from Mulliken, NPA or CHELPG method. It seems that other theoretical function besides the Coulomb attraction model should be introduced to discern whether path A is the most favorable nitration way. Here, the Hirshfeld charge is used to construct the condensed Fukui function (f) as well as condensed dual descriptor (Δf) according to the method presented by Fuentealba.47 The calculated f and Δf of the corresponding atoms are listed in Table S3.†
As shown in Table S3,† the f−(r) of C4 in A-IM1 is bigger than that in B-IM15 and Ca-IM5. Meanwhile, the Δf of C4 in A-IM13 is found to be more negative than that in B-IM15 and Ca-IM5. Based on the publication,47 such results indicate that C4 in A-IM13 is the most favorable sites to be attacked by NO2+. Hence path A must be the most likely nitration way, which is also in accordance with what were obtained from the Mulliken method. It is clear that the introduction order of –NO2 and the resulted fluctuation of charge distribution in the pre-intermediates do distinctly impact on the nitration of MDP.
While the impacts of the introduction order of –NO2 seem not so significant or dominant to promote the nitration if that of the HSO4− induction/enhancement are compared. The first NO2+ attacking step in path A is taken as an example (shown in Fig. 7 (insert)). As shown in Fig. 7, when MDP is directly nitrated/attacked by NO2+ (without any assistance of HSO4−) (Fig. S1†), the ΔG≠ (62.6 kcal mol−1) is much higher than that in the HSO4−-induced nitration of MDP (46.1 kcal mol−1), in which the HSO4−-induced H-transferring is not considered (Fig. S2†). However, when the HSO4−-induced H-transferring and H-abstracting are synergistically considered, the ΔG≠ in the NO2+ attacking step dramatically decreases to merely 5.6 kcal mol−1 (step ① in path A Fig. 2). Therefore, we strongly suggest that such HSO4−-induced nitration of MDP must be the most favorable way in the NO2+ attacking, and should be preferentially considered in this nitration system. We firmly believe that it is the incredible induction/enhancement of HSO4− that effectively promotes/catalyzes the nitration of MDP, and dramatically decreases the ΔG≠ of the titled nitration system.
3.2 Solvation effect on the nitration of MDP
Formamide as well as dimethylsulfoxide has been chosen as the model solvent to simulate the bulk solvent effects in the nitration of benzene with mixed acid via CPCM method.25 In the present work, the model solvents along with the CPCM method were also introduced and used to evaluate the solvent effects in the rate-determining step in paths A to C. The optimized geometries of the relevant species, and the corresponding energies (G and ΔG≠) in the gas (g), formamide (f) and dimethylsulfoxide (d) phases are shown in Fig. S7 and Table S4.†As shown in Fig. S7 and Table S4,† it is clear that though the energies of the relevant species for the rate-determining steps in paths A–C differ distinctly between the gas phase and the solvent phase (formamide or dimethylsulfoxide), the obtained optimized geometries of the relevant species and the ΔG≠ in every rate-determining step are quite analogous. It is indicated that the solvent effect in the nitration of MDP in nitric–sulfuric acids system may be inapparent or at least considerably weak to be omitted. We thus believed the presented mechanism for the titled nitration in the gas phase may be reliable and reasonable.
4. Conclusions
The HSO4− induced nitration mechanism of MDP with NO2+ to form N-MP is systematically investigated via B3LYP/6-311G(d,p) method. The possible nitration pathways, including the preferential dinitration on methyl group (paths A and A′), the preferential dinitration on methylene group (path B), and the crossed nitration (path C), are excavated and illustrated, respectively.A unique induction/enhancement of the co-existed HSO4− to the nitration is proposed and discerned. It is clearly indicated that HSO4−-induced nitration of MDP with NO2+ must be the most favorable way as compared with the nitration without the assistance of HSO4−. It is strongly suggested that the incredible induction/enhancement of HSO4− may effectively promote/catalyze the nitration of MDP with NO2+, including not only the NO2+ attacking, but the H-transferring and H-abstracting as well, and thus dramatically decrease the ΔG≠ of the nitration system.
The impact of the introduction order of –NO2 on the nitration is systematically demonstrated. Based on the ΔG≠ of the rate-determining step, it is found that path A must be more likely to occur, indicating that C10 in MDP may be preferentially dinitrated in the presence of HSO4−. Moreover, it is suggested that the NO2+ attacking may be dynamically affected by the introduced –NO2 and the resulted fluctuation of the charge distribution in the pre-intermediates. The atomic charge along with the Coulomb attractions between the corresponding atoms of the pre-intermediates is intentionally calculated, through which the reason why path A is the most favorable nitration way among all the possible paths is verified and discerned.
The solvent effect in the nitration of MDP in nitric–sulfuric acids seems to be inapparent. Reasonable mechanism may be proposed in the gas phase without considering the solvent effect.
We believe that the proposed HSO4−-induction, the fluctuation of the charge distribution and the resulted favorable nitration order may also fit for the similar nitration systems, such as the nitration of aromatics and other heterocyclic compounds. It is optimistically expected that the present study may provide a theoretical basis to the research and engineering tests of the preparation of FOX-7, and may strongly promote the exploration of other nitro energetic materials.
Acknowledgements
The authors gratefully acknowledge the financial support from the National Natural Science Foundation of China (21306111, 21327011), the Program for Changjiang Scholars and Innovative Research Team in University (IRT_14R33), the Shaanxi Innovative Team of Key Science and Technology (2013KCT-17), the Natural Science Research Program of Shaanxi Province (2014JM2034), and the Fundamental Research Funds for the Central Universities (GK201401001, GK201603103).Notes and references
- N. V. Latypov, J. Bergman, A. Langlet, U. Wellmar and U. Bemm, Tetrahedron, 1998, 54, 11525–11536 CrossRef CAS.
- N. V. Latypov, U. Wellmar and A. Langlet, US Pat., No. 6340780 B1, 2002.
- H. Q. Cai, Y. J. Shu, H. Huang, B. B. Cheng and J. S. Li, J. Org. Chem., 2004, 69, 4369–4374 CrossRef CAS PubMed.
- H. Q. Cai, Y. J. Shu, W. F. Yu, J. S. Li and B. B. Cheng, Acta Chim. Sin., 2004, 62, 295–301 CAS.
- A. K. Mandal, U. Thanigaivelan, R. K. Pandey, S. Asthana, R. B. Khomane and B. D. Kulkarni, Org. Process Res. Dev., 2012, 16, 1711–1716 CrossRef CAS.
- N. V. Latypov, M. Johansson, E. Holmgren, E. V. Sizova, V. V. Sizov and A. J. Bellany, Org. Process Res. Dev., 2007, 11, 56–59 CrossRef CAS.
- R. Gilardi, CCCD 127539, Cambridge Structural Database, Cambridge Crystallographic Data Center, Cambridge, U. K., 1999 Search PubMed.
- D. C. Sorescu, J. A. Boatz and D. L. Thopmson, J. Phys. Chem. A, 2001, 105, 5010–5021 CrossRef CAS.
- D. E. Taylor, F. Rob, B. M. Rice, R. Podeszwa and K. Szalewicz, Phys. Chem. Chem. Phys., 2011, 13, 16629–16636 RSC.
- (a) F. J. Zerilli and M. M. Kuklja, J. Phys. Chem. A, 2006, 110, 5173–5179 CrossRef CAS PubMed; (b) F. J. Zerilli and M. M. Kuklja, J. Phys. Chem. A, 2007, 111, 1721–1725 CrossRef CAS PubMed.
- (a) B. B. Averkiev, Z. A. Dreger and S. Chaudhuri, J. Phys. Chem. A, 2014, 118, 10002–10010 CrossRef CAS PubMed; (b) S. Hunter, P. L. Coster, A. J. Davidson, D. I. A. Millar, S. F. Parker, W. G. Marshall, R. I. Smith, C. A. Morrison and C. R. Pulham, J. Phys. Chem. C, 2015, 119, 2322–2334 CrossRef CAS.
- K. Z. Xu, J. R. Song, F. Q. Zhao, Z. H. Cao, H. X. Ma, R. Z. Hu, H. X. Gao and J. Huang, Acta Chim. Sin., 2007, 65, 2827–2831 CAS.
- G. Majano, S. Mintova, T. Bein and T. M. Klapötke, J. Phys. Chem. C, 2007, 111, 6694–6699 CAS.
- Q. Sun, Y. Zhang, K. Z. Xu, Z. Y. Ren, J. R. Song and F. Q. Zhao, J. Chem. Eng. Data, 2015, 60, 2057–2061 CrossRef CAS.
- J. Evers, T. M. Klapötke, P. Mayer, G. Oehlinger and J. Welch, Inorg. Chem., 2006, 45, 4996–5007 CrossRef CAS PubMed.
- X. Fang and W. G. McLuckie, J. Hazard. Mater., 2015, 285, 375–382 CrossRef CAS PubMed.
- W. A. Trzciński, S. Cudzilo, Z. Chylek and L. Szymańczyk, J. Hazard. Mater., 2008, 157, 605–612 CrossRef PubMed.
- (a) X. Z. Fan, J. Z. Li and Z. R. Liu, J. Phys. Chem. A, 2007, 111, 13291–13295 CrossRef CAS PubMed; (b) A. Gindulyté, L. Massa, L. Huang and J. Karle, J. Phys. Chem. A, 1999, 103, 11045–11051 CrossRef; (c) R. S. Bootha and L. J. Butlerb, J. Chem. Phys., 2014, 141, 134315/1–134315/9 Search PubMed; (d) V. G. Kiselev and N. P. Gritsan, J. Phys. Chem. A, 2014, 118, 8002–8008 CrossRef CAS PubMed; (e) Y. Liu, F. Li and H. Sun, Theor. Chem. Acc., 2014, 133, 1567/1–1567/11 Search PubMed; (f) B. Yuan, Z. J. Yu and E. R. Bernstein, J. Chem. Phys., 2014, 140, 074708/1–074708/9 CAS; (g) H. X. Gao, F. Q. Zhao, R. Z. Hu, Q. N. Pan, B. Z. Wang, X. W. Yang, Y. Gao, S. L. Gao and Q. Z. Shi, Chin. J. Chem., 2006, 24, 177–181 CrossRef CAS.
- G. Hervé, G. Jacob and N. Latypov, Tetrahedron, 2007, 63, 953–959 CrossRef.
- H. X. Gao and J. M. Shreeve, Angew. Chem., Int. Ed., 2015, 54, 6335–6338 CrossRef CAS PubMed.
- (a) E. V. Sizova, V. V. Sizov and D. I. V. Tselinskii, Russ. J. Org. Chem., 2007, 43, 1232–1237 CrossRef CAS; (b) T. T. Vo and J. M. Shreeve, J. Mater. Chem. A, 2015, 3, 8756–8763 RSC.
- E. K. Kim, T. M. Bockman and J. K. Kochi, J. Am. Chem. Soc., 1993, 115, 3091–3104 CrossRef CAS.
- P. M. Esteves, J. W. D. M. Carneiro, S. P. Cardoso, A. G. H. Barbosa, K. K. Laali, G. Rasul, G. K. Surya Prakash and G. A. Olah, J. Am. Chem. Soc., 2003, 125, 4836–4849 CrossRef CAS PubMed.
- V. D. Parker, T. Kar and D. Bethell, J. Org. Chem., 2013, 78, 9522–9525 CrossRef CAS PubMed.
- G. Koleva, B. Galabov, B. Hadjieva, H. F. Schaefer III and P. V. R. Schleyer, Angew. Chem., Int. Ed., 2015, 54, 14123–14127 CrossRef CAS PubMed.
- Z. H. Zhen and Y. R. Mo, J. Chem. Theory Comput., 2013, 9, 4428–4435 CrossRef PubMed.
- L. T. Chen, H. M. Xiao, J. J. Xiao and X. D. Gong, J. Phys. Chem. A, 2003, 107, 11440–11444 CrossRef CAS.
- L. T. Chen, H. M. Xiao and J. J. Xiao, J. Phys. Org. Chem., 2005, 18, 62–68 CrossRef CAS.
- L. X. Jin, L. Wang, C. Y. Zhang, W. L. wang, S. T. Min and D. D. Hu, Phys. Chem. Chem. Phys., 2014, 16, 16264–16267 RSC.
- K. Wang, J. G. Chen, B. Z. Wang, F. Y. Liu, Z. T. Liu, Z. W. Liu, W. L. Wang, J. Q. Jiang, Z. P. Hao and J. Lu, RSC Adv., 2015, 5, 25183–25191 RSC.
- K. Wang, J. G. Chen, B. Z. Wang, J. Lu, W. L. Wang, F. Y. Liu, C. Zhou, P. Lian, Z. W. Liu and Z. T. Liu, Chem. J. Chin. Univ., 2015, 36, 531–538 CAS.
- The hydrolysis of N-MP to form FOX-7 will be presented elsewhere.
- N. C. Marziano, A. Tomasin, C. Tortato and J. S. Zaldivar, J. Chem. Soc., Perkin Trans. 2, 1998, 2, 1973–1982 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision B.01, Gaussian, Inc, Wallingford, CT, 2010 Search PubMed.
- C. Lee, W. T. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- A. D. McLean and G. S. Chandler, J. Chem. Phys., 1980, 72, 5639–5648 CrossRef CAS.
- C. Gonzales and H. B. Schlegel, J. Chem. Phys., 1989, 90, 2154–2161 CrossRef.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104–154119 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, J. Comput. Chem., 2011, 32, 1456–1465 CrossRef CAS PubMed.
- R. S. Mulliken, J. Chem. Phys., 1955, 23, 1833–1840 CrossRef CAS.
- (a) V. Barone and M. Cossi, J. Phys. Chem. A, 1998, 102, 1995–2001 CrossRef CAS; (b) M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681 CrossRef CAS PubMed.
- (a) F. Cacace, M. Attinà, G. D. Petris and M. Speranza, J. Am. Chem. Soc., 1990, 112, 1014–1018 CrossRef CAS; (b) E. D. Hughes, C. Ingold and R. B. Pearson, J. Chem. Soc., 1958, 4357–4365 RSC; (c) E. L. Blackall, E. D. Hughes, C. Ingold and R. B. Pearson, J. Chem. Soc., 1958, 4366–4374 RSC; (d) T. J. Lee and J. E. Rice, J. Phys. Chem., 1992, 96, 650–657 CrossRef CAS.
- A. E. Reed, R. B. Weinstock and F. Weinhold, J. Chem. Phys., 1985, 83, 735–746 CrossRef CAS.
- A. E. Reed, L. A. Curtiss and F. Weinhold, Chem. Rev., 1988, 88, 899–926 CrossRef CAS.
- C. M. Breneman and K. B. Wiberg, J. Comput. Chem., 1990, 11, 361–373 CrossRef CAS.
- F. L. Hirshfeld, Theor. Chim. Acta, 1977, 44, 129–138 CrossRef CAS.
- P. Fuentealba, E. Florez and W. Tiznado, J. Chem. Theory Comput., 2010, 6, 1470–1478 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The calculated Mulliken atomic charges of MDP and the relevant intermediate. Schematic energy diagram of the direct nitration of MDP with NO2+. Schematic energy diagram of the HSO4−-induced nitration of MDP with NO2+. Schematic energy diagram of the direct H-transfer of mono-nitro substitution product. Schematic energy diagram of the direct H-transfer of di-nitro substitution product. Schematic energy diagram of the trans-HSO4−-induced nitration in the second nitration step in path C. Free energies and active free energies for the nitration of MDP with NO2+ in the gas, formamide and dimethylsulfoxide phases. Optimized geometries of species for the rate-limiting step in paths A–C. See DOI: 10.1039/c6ra18842g |
| This journal is © The Royal Society of Chemistry 2016 |