
The enhanced performance of a CeSiOx support on a Mn/CeSiOx catalyst for selective catalytic reduction of NOx with NH3
Wei Liab,
Rui-tang Guo*ab,
Shu-xian Wangab,
Wei-guo Panab,
Qi-lin Chenab,
Ming-yuan Liab,
Peng Sunab and
Shu-ming Liuab
aSchool of Energy Source and Mechanical Engineering, Shanghai University of Electric Power, Shanghai, P. R. China
bShanghai Engineering Research Center of Power Generation Environment Protection, Shanghai, P. R. China. E-mail: grta@zju.edu.cn
First published on 26th August 2016
Abstract
A series of Mn/CeSiOx catalysts were prepared by the wet impregnation method and used for selective catalytic reduction of NO with NH3. As can be seen from the experimental results, the Mn/CeSiOx catalyst with a Ce/Si molar ratio of 2/1 showed excellent low-temperature SCR activity, high N2 selectivity and excellent SO2 and H2O tolerance. The relationship between the CeSiOx support and the SCR performance of Mn/CeSiOx (2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) catalyst was investigated based on the characterization results of N2 adsorption, XRD, XPS, H2-TPR, NH3-TPD and in situ DRIFT. The strong interaction between Ce and Si resulted in the good dispersion of Mn species on the support; correspondingly, the redox ability and NH3 adsorption capacity were greatly enhanced. The results of in situ DRIFT study revealed that the NH3-SCR reactions over Mn/CeO2 and Mn/CeSiOx (2:1) mainly obeyed both the E–R mechanism and the L–H mechanism. Furthermore, the formation of more Mn4+ and chemisorbed oxygen greatly facilitates the oxidation of NO to NO2, as a result, promoting the low-temperature SCR performance of Mn/CeSiOx (2:1).
:1) catalyst was investigated based on the characterization results of N2 adsorption, XRD, XPS, H2-TPR, NH3-TPD and in situ DRIFT. The strong interaction between Ce and Si resulted in the good dispersion of Mn species on the support; correspondingly, the redox ability and NH3 adsorption capacity were greatly enhanced. The results of in situ DRIFT study revealed that the NH3-SCR reactions over Mn/CeO2 and Mn/CeSiOx (2:1) mainly obeyed both the E–R mechanism and the L–H mechanism. Furthermore, the formation of more Mn4+ and chemisorbed oxygen greatly facilitates the oxidation of NO to NO2, as a result, promoting the low-temperature SCR performance of Mn/CeSiOx (2:1).
Introduction
Several environmental problems such as acid rain, photochemical smog and haze are closely related to the emission of NOx from the combustion process of fossil fuels from stationary sources and mobile sources.1–3 In the past several decades, several techniques have been developed and applied for the control of NOx emission. Among them, the selective catalytic reduction of NOx with NH3 using vanadium-based catalyst has been put into industrial application for reducing NOx from stationary sources such as coal-fired power plants for about 40 years.4 This catalyst exhibits high activity in the narrow temperature window of 300–400 °C.5 However, several inevitable drawbacks are still associated with it including the toxicity of vanadium species, high conversion of SO2 to SO3 and the deactivation by alkali metals in the fly ash.6–10 Therefore, it is of great demand to develop vanadium-free catalysts with high activities at low temperatures.In recent years, manganese-based catalyst has received much attention due to the presence of various types of labile oxygen in MnOx, which could promote the catalytic cycle in SCR reactions.11,12 In previous studies, MnOx supported on different carriers such as TiO2,13,14 Al2O3,15 carbon nanotube12,16 and palygorskite17 exhibited excellent low-temperature SCR performances. However, the deactivation of manganese-based catalyst by SO2 is still a barrier to its industrial application.18 It is generally accepted that the support has a crucial impact on the performance of SCR catalyst. For the past few years, some carriers based on composite oxides such as TiO2–Al2O3,19 CeO2–ZrO2,20–22 TiO2–SiO2,23 CeTiOx (ref. 24) and TiWOx (ref. 25) have been successfully applied as the support of SCR catalysts to enhance their SCR performances. Therefore, MnOx catalyst supported on composite oxides may be a competitive candidate for future industrial application. In this study, Mn/CeSiOx catalyst was synthetized and used in NH3-SCR reaction. The experimental results revealed that the Mn/CeSiOx catalyst with a Ce/Si molar ratio of 2/1 showed excellent low-temperature SCR activity, high N2 selectivity and excellent SO2 and H2O tolerance. The promotion mechanism of CeSiOx support on its SCR performance was also investigated based on the characterization results.
Experimental
All chemicals used in this study were of analytical grade and supplied by Aladdin Reagent Inc., China. Firstly, the CeSiOx support was prepared by citric acid method. Cerium nitrate (CN), tetraethyl orthosilicate (TEOS) and citric acid (CA) were mixed in the desired molar proportions, and the mole ratio of CA/(Si + Ce) was set as 1.0 (Ce/Si molar ratio = 2:1; 1:1 and 1:2 respectively). After stirred at room temperature for 1 h, the mixture was dried at 100 °C for 12 h, a foam-like solid would come into being. Then the foam-like solid was calcined in air at 500 °C for 5 h to obtain the final CeSiOx support. Similarly, the pure CeO2 and SiO2 supports were prepared by using CN and TEOS as the precursors of Ce and Si respectively.
The Mn/CeSiOx catalyst was prepared by the impregnation method. Firstly, the CeSiOx support (3 mL) was impregnated by incipient wetness with an aqueous solution of manganese nitrate (the mass ratio of Mn/CeSiOx = 0.15). After that, the sample was dried at 100 °C for 12 h, followed by calcination in air at 500 °C for 5 h. Based on the same method, Mn/CeO2 and Mn/SiO2 catalyst samples with a Mn/support mass ratio of 0.15 were prepared and used as the counterparts. For convince, the Mn/CeSiOx catalyst was denoted as Mn/CeSiOx(y), where y represents the molar ratio between Ce and Si.
Nitrogen adsorption measurements were performed on a Quantachrome Autosorb-iQ-AG instrument based on nitrogen gas sorption at 77 K and analyzed by Quantachrome AS1Win software (Quantachrome Instruments Version 2.01). Prior to the experiments, the samples were heated to 200 °C under vacuum for 1 h. The specific surface area was evaluated by the Brunauer–Emmett–Teller (BET) method. And the pore volumes and average pore diameters were determined by the Barrett–Joyner–Halenda (BJH) method from the desorption branches of the isotherms.
To determine the crystal structures of the catalyst samples, X-ray powder diffraction patterns were collected by a Bruker D8 Advance diffractometer using a graphite monochrometer with Cu Kα radiation (λ = 0.15406 nm). The 2θ scans cover the range of 20–80°.
To evaluate the chemical states for all the elements on the surfaces of the catalyst samples, X-ray photoelectron spectroscopy was performed on a Thermo ESCALAB 250 spectrometer. An Al Kα monochromatized radiation (hν = 1486.6 eV) was employed as the X-ray source. Survey spectra were recorded with a pass energy of 100 eV and high resolution spectra of the XPS O 1s–V 2p, C 1s and valence band region were recorded with a pass energy of 20 eV. The shift of the binding energy was calibrated on the basis of C 1s level at 284.8 eV.
Raman spectra were recorded on a Renishaw inVia Raman Microscope (Renishaw, Britain) with Ar+ radiation (514 nm) and the laser power of 20 mW.
Temperature-programmed reduction of H2 (H2-TPR) and temperature-programmed desorption of NH3 (NH3-TPD) were carried out on an Autosorb-iQ-C TPR/TPD automatic chemisorption analyzer (Quantachrome Instruments, USA) using 50 mg catalyst samples. Prior to the TPR experiments, the catalyst sample was pretreated at 400 °C in He for 1 h. Then the TPR runs were carried out with linear heating rate (10 °C min−1) up to 700 °C in pure Ar containing 5% H2 at a flow rate of 40 mL min−1. For NH3-TPD experiments, the samples were also pretreated in He at 400 °C for 1 h. Then the samples were purged under 500 ppm NH3 at a flow rate of 30 mL min−1 for 30 min. Then the desorption was carried out by heating the catalyst sample in He (30 mL min−1) form 100 to 750 °C. The signal of H2 or NH3 was recorded by a thermal conductivity detector (TCD).
The in situ DRIFT measurements were performed on a FTIR spectrometer (Nicolet iS50, Thermo, USA) equipped with a smart collector and an MCT/A detector cooled by liquid nitrogen. In the DRIFT cell (ZnSe window) connected with a gas flow system, the samples preheated at 500 °C for 2 h in N2 flow and cooled down to 200 °C to get a background spectrum. Then the background spectra were automatically subtracted from the sample spectrum. During the in situ DRIFT measurements, the gas mixture contains 500 ppm NH3, or/and 500 ppm NO + 5% O2, balance N2, with a flow rate of 300 mL min−1. All the spectra were recorded by accumulating 100 scans with a resolution of 4 cm−1. The infrared spectra in the region of 1100–2000 cm−1 were recorded.
The SCR activities of the catalyst samples were measured at atmospheric pressure in a fixed-bed continuous flow quartz reactor (i.d. = 8 mm). In each experimental run, about 0.55 mL catalyst sample was used. The reactant gas contains 600 ppm NH3, 600 ppm NO, 5% O2, 100 ppm SO2 (when used), 5% H2O (when used) and balance Ar, with a total flow rate of 1 L min−1. So the gas hourly space velocity (GHSV) was about 108000 h−1. The concentrations of NO and NO2 in the effluent gas stream were monitored by a Thermo NO-NOx chemiluminescence analyzer (Model 42i-HL). The concentration of NH3 in the outlet gas stream was measured by a NH3 analyzer (Model 17i). And the concentration of N2O was analyzed by a Nicolet iS50 spectrometer equipped with a gas cell with 0.2 L volume.
After the SCR reaction reached a steady state, the values of NOx conversion and N2 selectivity could be obtained accordingly:26,27
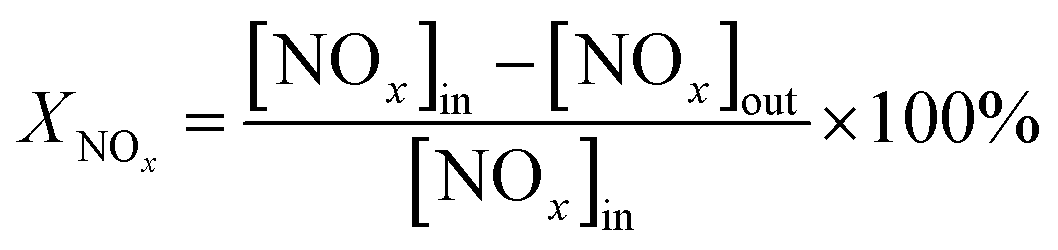 | (1) |
 | (2) |
Results and discussion
As show in Fig. 1(A), the catalyst samples supported on the composite oxides exhibit higher SCR activity than Mn/SiO2 and Mn/CeO2, especially in the low temperature range (<250 °C). It seems that the molar ratio of Ce/Si has a great impact on the SCR performance of Mn/CeSiOx catalyst, and Mn/CeSiOx (2:1) catalyst shows more than 90% NOx conversion in the temperature range of 150–300 °C under a GHSV of 108000 h−1. Thus the modification of CeO2 support by Si could greatly enhance the low-temperature SCR activity of Mn/CeSiOx (2:1) catalyst. Combined with the results of N2 selectivity (Fig. 1(B)), it can be observed that the composite oxides could also enhance the selectivity; moreover, Mn/CeSiOx (2:1) catalyst is of the highest N2 selectivity among the five catalyst samples. Therefore, we focus on Mn/CeSiOx (2:1) catalyst in the following study.
 | ||
| Fig. 1 (A) NOx conversions over the five catalyst samples as a function of reaction temperature. (B) N2 selectivities over the five catalyst samples as a function of reaction temperature. (C) Normalized reaction rate of NO by BET surface area as a function of reaction temperature. Reaction conditions: [NO] = [NH3] = 600 ppm, [O2] = 5%, balance Ar, GHSV = 108000 h−1. | ||
It is well known that the BET surface of SCR catalyst has a great impact on its catalytic performance. To eliminate the effect of specific surface area on the SCR performance, the reaction rate normalized by BET surface area was calculated by the following equation:28,29
| RNOx = XNOxQCf/(WSBET) | (3) |
The results of NO reaction rate normalized by BET surface area are shown in Fig. 1(C). As can be seen from Fig. 1(C), the trend of normalized reaction rate is similar with that of NOx conversions. The reaction rates of catalysts supported on the composite oxides are higher than that of Mn/CeO2 and Mn/SiO2, and Mn/CeSiOx (2:1) exhibits the highest reaction rate among the five catalyst samples.
The deactivation effect of SO2 and H2O on SCR catalyst has been extensively reported in many studies.30–34 As presented in Fig. 2, the NOx conversion over Mn/CeSiOx (2:1) at 250 °C only decreases a little after the introduction of 100 ppm SO2 and 5% H2O. In addition, the NO conversion over Mn/CeSiOx (2:1) recovers to a great extent after the cut of SO2 and H2O. On the contrary, the NOx conversion over Mn/CeO2 decreases a lot in the presence of 100 ppm SO2 and 5% H2O, and the SCR activity is nearly unrecoverable after the cut of SO2 and H2O. Combined with its good SCR performance and high resistance to SO2 and H2O, it can be seen that Mn/CeSiOx (2:1) is a promising deNOx catalyst for future application.
 | ||
| Fig. 2 Effect of SO2 and H2O on the NOx conversions over Mn/CeSiOx (2:1) and Mn/CeO2 catalysts at 250 °C. Reaction conditions: [NO] = [NH3] = 600 ppm, [SO2] = 100 ppm, [O2] = [H2O] = 5%, balance Ar, GHSV = 108000 h−1. | ||
The results of BET analysis are summarized in Table 1. It is clear that the catalyst samples supported on composite oxides exhibit higher specific surface areas than Mn/CeO2 and Mn/SiO2, which should be originated from the strong interaction between Ce and Si in the support. The enhanced BET surface areas of the CeSiOx-supported catalyst samples are favorable to the adsorption of reactants on them and facilitate the NH3-SCR reaction. On the contrary, the pore volumes of Mn/CeO2 and Mn/SiO2 are larger than that of the CeSiOx-supported catalyst samples, indicating that the effect of pore volume on BET surface area is very little.
| Samples | BET surface area (m2 g−1) | Average pore diameter (nm) | Pore volume (cm3 g−1) | Mean crystallite size of CeO2 (nm) |
|---|---|---|---|---|
| Mn/CeSiOx (2:1) |
89.14 | 0.155 | 11.15 | 8.0 |
| Mn/CeSiOx (1:1) |
84.60 | 0.150 | 12.34 | 10.1 |
| Mn/CeSiOx (1:2) |
78.53 | 0.141 | 12.09 | 11.2 |
| Mn/CeO2 | 58.09 | 0.121 | 16.05 | 16.8 |
| Mn/SiO2 | 66.34 | 0.133 | 14.47 | — |
The XRD patterns of the five catalyst samples are illustrated in Fig. 3. Noticeably, only the diffraction peaks of CeO2 and SiO2 could be detected, suggesting that Mn species in the catalyst samples are presented in amorphous structure. For Mn/SiO2, only a very weak peak of SiO2 could be observed, therefore, its crystallinity is very low. However, its SCR activity is not very high, which may be due to the participation of Ce species in the NH3-SCR reaction for other samples. Mn/CeO2 is of the highest crystallinity among the five catalyst samples, corresponding to the lowest SCR activity (Fig. 1(A)). For the catalyst samples with a composite oxides, their crystallinities are in the following order: Mn/CeSiOx (2:1) < Mn/CeSiOx (1:1) < Mn/CeSiOx (1:2). Therefore, the strong interaction between Ce and Si could hinder the formation of CeO2 crystal, leading to the decrease of its peak intensity. Furthermore, lower ceria crystallinity means the presence of more lattice defects, which is favorable to the formation of Ce3+.35 The mean particle sizes of CeO2 in different catalyst samples were also calculated by the Scherrer equation and the results are listed in Table 1. It can be seen that the crystallite size of CeO2 in Mn/CeSiOx (2:1) is much smaller than that in other catalyst samples, suggesting the presence of more lattice defects in Mn/CeSiOx (2:1), agreeing well with the results mentioned above.
 | ||
| Fig. 3 XRD patterns of the five catalyst samples. | ||
Compared with XRD analysis, Raman spectra are more surface sensitive for the characterization of the different catalyst samples. As shown in Fig. 4, the main band at about 463 cm−1 could be assigned to the F2g vibration mode of the cubic fluorite structure, which is due to the an octahedral local symmetric breathing mode of oxygen atoms around the ceria lattice.36 As can be observed from Fig. 4, the characteristic F2g vibration modes of the three Mn/CeSiOx catalyst samples are weakened, broadened and shifted to lower frequency, indicating the presence of reduced state of cerium.37,38
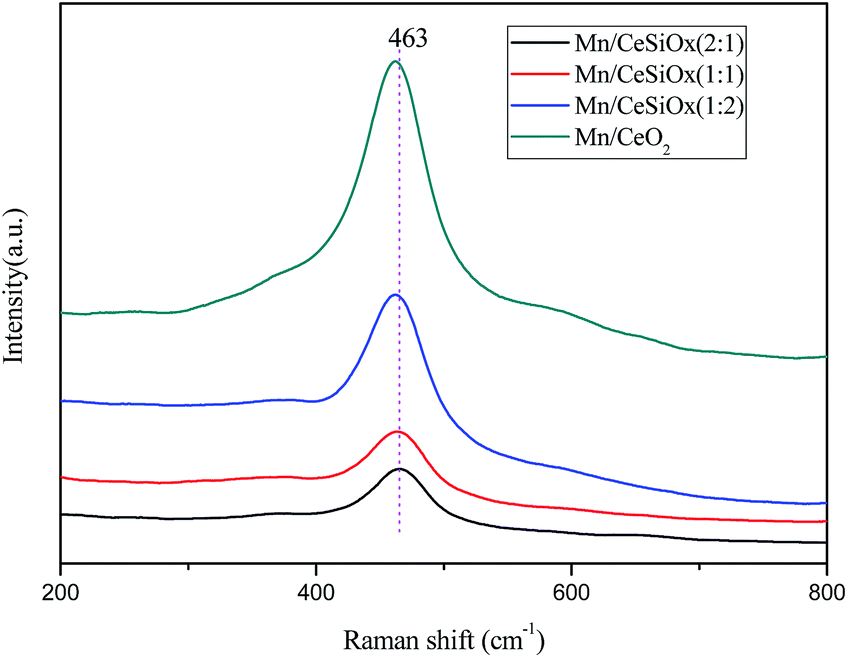 | ||
| Fig. 4 Raman spectra of different catalyst samples. | ||
The chemical states of Mn, Ce and O species on Mn/CeO2 and Mn/CeSiOx (2:1) are determined by XPS analysis, and the results are shown in Fig. 5.
 | ||
| Fig. 5 XPS spectra of the two catalyst samples: (A) Mn 2p3/2; (B) Ce 3d; (C) Si 2p; (D) O 1s. | ||
As presented in Fig. 5(A), the XPS spectra of Mn 2p3/2 contain three peaks after a peak-fitting deconvolution, which could be assigned to Mn2+, Mn3+ and Mn4+ respectively.39–41 On the basis of XPS analysis results, the values of Mn4+ concentrations over the two catalyst samples could be calculated, as summarized in Table 2. It is noticeable that the Mn4+ concentration over Mn/CeSiOx (2:1) is much higher than that over Mn/CeO2. According to previous studies, species with higher oxidation state are more active for redox reactions over manganese-based catalysts.42 And the study of Liu et al.43 also indicated that the enhanced NO oxidation to NO2 by Mn4+ could greatly promote the NH3-SCR reaction. Therefore, the relatively higher surface Mn4+ may partly contribute to the high low-temperature SCR activity of Mn/CeSiOx (2:1).
| Samples | Mn (at%) | Ce (at%) | O (at%) | Mn4+ (at%) | Ce3+ (at%) | Oβ (at%) |
|---|---|---|---|---|---|---|
| Mn/CeO2 | 4.43 | 16.70 | 78.87 | 1.19 | 3.30 | 27.57 |
| Mn/CeSiOx (2:1) |
4.68 | 13.94 | 76.08 | 1.67 | 3.64 | 29.07 |
The XPS spectra of Ce 3d for the two catalyst samples are illustrated in Fig. 5(B). It contains two multiplets corresponding to the spin orbit split 3d5/2 and 3d3/2 core holes respectively. According to previous studies,44–46 the peaks denoted as u, u2, u3, v, v2 and v3 could be assigned to Ce4+, while the two peaks denoted as u1 and v1 could be ascribed to Ce3+. According to the XPS spectra of Ce 3d, Ce4+ and Ce3+ are concomitant on the surfaces of Mn/CeO2 and Mn/CeSiOx (2:1). Moreover, the Ce3+ concentration on Mn/CeSiOx (2:1) is higher than that over Mn/CeO2. It is well recognized that Ce3+ is originated from the defects of ceria defects, so the results of XPS analysis agree well with that of XRD analysis. Based on previous studies,35,47 Ce3+ could form oxygen vacancies and unsaturated chemical bonds, facilitating the generation of chemisorbed oxygen on catalyst surface, which will be discussed in the following sections.
The XPS spectra of Si 2p of Mn/CeO2 and Mn/CeSiOx (2:1) are shown in Fig. 5(C). As can be seen from Fig. 5(C), the binding energy of Si 2p in the XPS spectra of Mn/SiO2 is at 103.4 eV, which is in accordance with the results of Hu et al.48 For Mn/CeSiOx (2:1), the binding energy of Si 2p in the spectra of Mn/CeSiOx (2:1) is lower than that in the spectra of Mn/CeO2, indicating the formation of Ce–O–Si bond. And the charge disbalance formed in this process is favorable to the generation of acid sites,49 as proven by the results of NH3-TPD analysis.
Furthermore, the chemical states of oxygen species on the two catalyst samples were investigated by XPS analysis, as shown in Fig. 5(D). Obviously, the O 1s spectra could be fitted into two peaks: the lattice oxygen (529.0–530.0 eV, denoted as Oα) and the chemisorbed oxygen (531.3–531.9 eV, denoted as Oβ).50–52 From Table 2, it could be seen that the concentration of Oβ over Mn/CeSiOx (2:1) is higher than that over Mn/CeO2, in good accordance with the different Ce3+ concentrations on them. As an active oxygen species in NH3-SCR reaction, chemisorbed oxygen is of high mobility and could easily exchange with the oxygen in the gas or in the molecules adsorbed on catalyst, as a result, promoting the oxidation of NO to NO2, accompanied by the enhanced “fast SCR” reaction.12,53,54 According to the study of Chen et al.,55 the oxidation of NO to NO2 was of great importance to the SCR reaction below 250 °C, so the enrichment of Mn4+ and chemisorbed oxygen on the surface of Mn/CeSiOx (2:1) by the introduction of Si is crucial to its excellent low-temperature SCR performance.
H2-TPR analysis was performed to investigate the redox behavior of the catalyst samples. As can be seen from Fig. 6, each TPR profile contains two reduction peaks. Based on the results of XRD and TPR, the first peak in the profile of Mn/CeSiOx (2:1) at about 345 °C could be attributed to the reduction of Mn4+ to Mn3+, and the second one at about 461 °C may be ascribed to Mn3+ to Mn2+ and the reduction of amorphous CeO2 on catalyst surface.35,56–58 So the results of H2-TPR analysis are in good accordance with the results of XPS analysis. The reduction of bulk CeO2 is neglected because it happens above 750 °C.59 For Mn/CeO2 and Mn/SiO2, the reduction peaks in their TPR profiles could also be assigned to Mn4+ → Mn3+ and Mn3+ → Mn2+ respectively. Compared with Mn/CeSiOx (2:1), the reduction temperatures of Mn/CeO2 and Mn/SiO2 move to higher values, suggesting that the redox abilities of Mn/CeO2 and Mn/SiO2 are lower than that of Mn/CeSiOx (2:1). Furthermore, larger reduction peaks of Mn/CeSiOx (2:1) also indicates the presence of more active oxygen than the other two catalyst samples. It is generally accepted that the reducing ability of Mn-based catalyst plays an important role to complete the catalytic cycle in SCR reaction.60 So the high redox ability of Mn/SiOx (2:1) should also be responsible for its excellent SCR performance.
 | ||
| Fig. 6 H2-TPR profiles of the three catalyst samples. | ||
It is well recognized that the adsorption and activation of NH3 species is a key step for NH3-SCR reaction.61 Therefore, NH3-TPD technique was used to study the surface acid properties of the catalyst samples. As shown in Fig. 7, there is a broad desorption peak could be observed in the profiles of Mn/CeO2 and Mn/SiO2 from 100–500 °C, suggesting the presence of acid sites with different thermal stabilities on them. For Mn/CeSiOx (2:1), there are three desorption peaks located at 137, 202 and 364 °C respectively, which could be related to the desorption of physisorbed NH3 and some NH4+ bounded to the weak Brønsted acid sites, NH4+ from strong Brønsted acid sites and the coordinated NH3 bound to the Lewis acid sites, respectively.62,63 Moreover, the total desorption peak area of Mn/CeSiOx (2:1) is much larger than that of Mn/CeO2 and Mn/SiO2, indicating the presence of more acid sites on its surface. Therefore, the composite oxide support could provide more acid sites for NH3 adsorption.
 | ||
| Fig. 7 NH3-TPD profiles of the three catalyst samples. | ||
To investigate the NH3-SCR reaction mechanism over the catalyst samples, in situ DRIFT study was performed to identify the active sites, adsorbed species and intermediates under different conditions.
To determine the surface acid properties of the catalyst samples, the in situ DRIFT spectra of NH3 adsorption over Mn/CeO2 and Mn/CeSiOx (2:1) at 200 °C were recorded, and the results are shown in Fig. 8. As can be seen from Fig. 8, several bands could be observed. The band at 1710 and 1433 cm−1 could be assigned to NH4+ species on Brønsted acid sites, and the bands at 1617, 1258 and 1156 cm−1 could be attributed to NH3 species adsorbed on Lewis acid sites.64–67 The band at 1533 cm−1 belongs to NH2 species, as an important immediate in NH3-SCR reaction, which could react with NO to form N2 and H2O.68 While this band is not found in the spectra of Mn/CeO2. It is noticeable that the intensities of the bands in the spectrum of Mn/CeSiOx (2:1) are higher than that in the spectrum of Mn/CeO2, suggesting more NH3 species are adsorbed on the surface of Mn/CeSiOx (2:1), as also reflected by the results of NH3-TPD analysis.
 | ||
| Fig. 8 In situ DRIFT spectra of NH3 adsorption over the two catalyst samples at 200 °C. | ||
The in situ DRIFT spectra of NO + O2 co-adsorption over the two catalyst samples are shown in Fig. 9. The bands at 1644 cm−1 could be assigned to bridged nitrate; the band at 1575, 1563 and 1239 cm−1 could be corresponded to bidentate nitrate; and the band at 1392 and 1284 cm−1 should be originated from monodentate nitrate species.69–71 Due to the presence of more acid sites on Mn/CeSiOx (2:1), it shows less NOx species adsorption amount than Mn/CeO2.71
 | ||
| Fig. 9 In situ DRIFT spectra of NO + O2 co-adsorption over the two catalyst samples at 200 °C. | ||
To identify the role of adsorbed NH3 species in the NH3-SCR reactions over the catalyst samples, the in situ DRIFT spectra of the reaction between the preadsorbed NH3 species and NO + O2 at 200 °C as a function of time were recorded, as shown in Fig. 10. From Fig. 10(A), two bands of adsorbed NH3 species (1700 and 1533 cm−1) in the spectra of Mn/CeO2 could be observed after the exposure of NH3. And these bands disappeared quickly after the introduction of NO + O2, indicating that all the adsorbed NH3 species are active in the NH3-SCR reaction over Mn/CeO2 catalyst. Meanwhile, two bands (1566 and 1256 cm−1) of adsorbed NOx species appeared and increased with time. As for Mn/CeSiOx (2:1), a similar trend could also be observed in its DRIFT spectra of the reaction between NO + O2 and preadsorbed NH3 species. Compared with Fig. 10(A) and (B), it seems that the adsorbed NH3 species on Mn/CeSiOx (2:1) is more active than that absorbed on Mn/CeO2. For example, the band at 1533 cm−1 in Fig. 10(A) disappeared in about 5 minutes, while the band at 1433 cm−1 vanished in about 2 minutes. Thus the composite oxides support may have an activation effect on adsorbed NH3 species. The quick disappearance of adsorbed NH3 species after the introduction of NO + O2 indicates the existence of E–R mechanism.72
 | ||
| Fig. 10 In situ DRIFT spectra of the reaction between NO + O2 and preadsorbed NH3 species over: (A) Mn/CeO2; (B) Mn/CeSiOx (2:1) at 200 °C. | ||
On the other hand, the in situ DRIFT spectra of the reaction between NH3 and preadsorbed NOx species are shown in Fig. 11. As presented in Fig. 11(A), several bands of adsorbed NOx species (1569, 1398 and 1240 cm−1) were detected in the spectra of Mn/CeO2 after the exposure of NO + O2. After NH3 was passed over the catalyst, the two bands at 1569 and 1240 cm−1 vanished in about 2 minutes, while the band at 1398 cm−1 kept rarely changed in the whole process. Thus only the bidentate nitrate is active in the NH3-SCR reaction over Mn/CeO2 catalyst, while the monodentate nitrate is a spectator in this process. Furthermore, several bands of adsorbed NH3 species formed with the introduction of NH3. For Mn/CeSiOx (2:1), a similar phenomenon could also be observed (Fig. 11(B)), only the bidentate nitrate could participate in the NH3-SCR reaction over it. Therefore, the adsorbed NOx species are of different activities in the NH3-SCR reaction, which is also reported by Liu et al.65 Combined the results of in situ DRIFT study, we may conclude that the adsorbed NH3 species and the bidentate nitrate species are all active in the NH3-SCR reactions over the two catalyst samples, thus the L–H mechanism is also applicable for them.
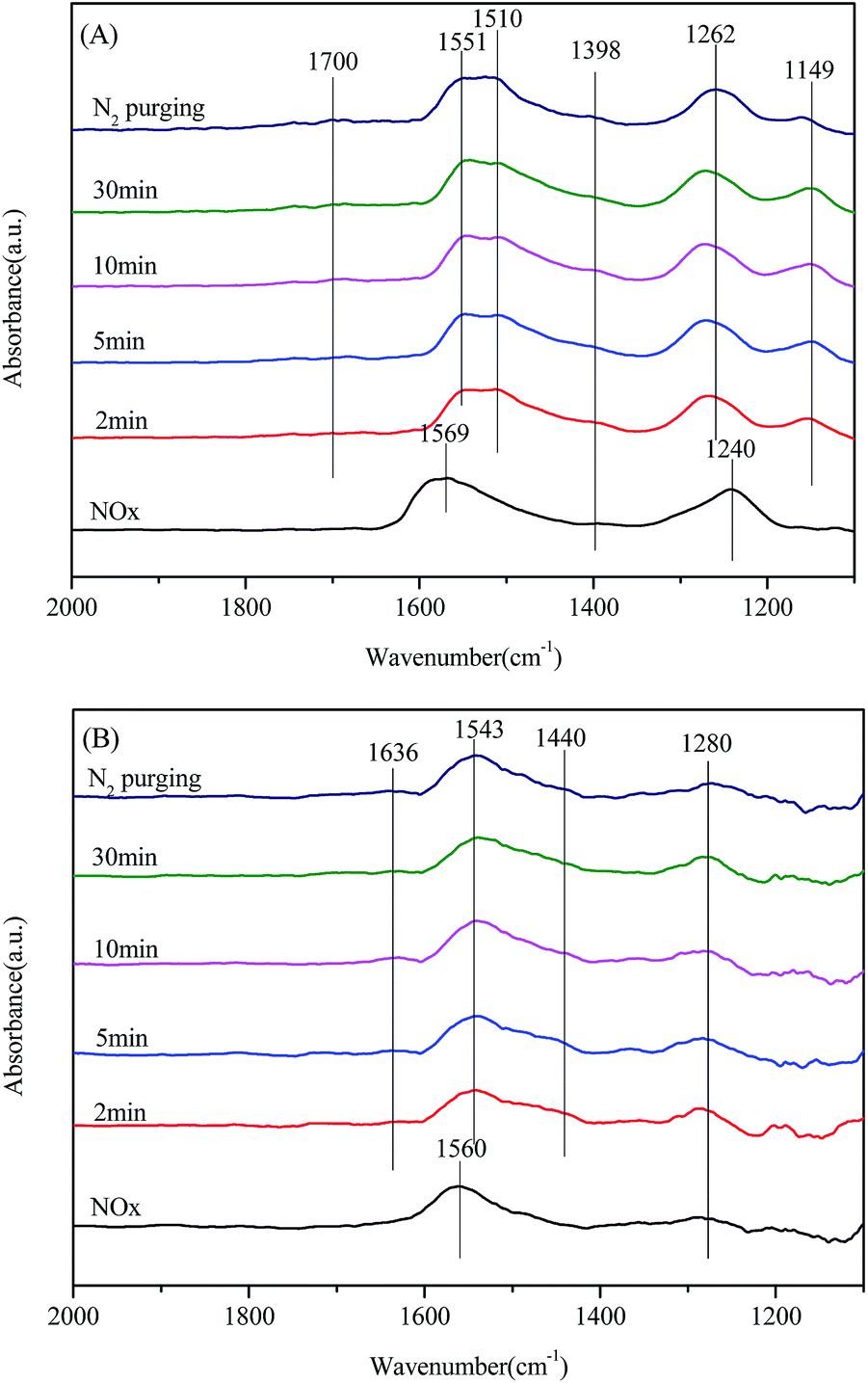 | ||
| Fig. 11 In situ DRIFT spectra of the reaction between NH3 and preadsorbed NOx species over: (A) Mn/CeO2; (B) Mn/CeSiOx (2:1) at 200 °C. | ||
Conclusions
Mn/CeSiOx (2:1) catalyst shows higher low-temperature SCR activity and better SO2 and H2O resistance than Mn/CeO2 catalyst. From the characterization results, it was found that the modification of CeO2 support of Mn/CeO2 catalyst by Si leads to lower crystallinity, more reducible species and acid sites, which are all crucial to the NH3-SCR reaction over it. The results of in situ DRIFT study reveal the coexistence of E–R mechanism and L–H mechanism. And the presence of more Mn4+ and chemisorbed oxygen should play a primary role for the greatly enhanced low-temperature SCR performance of Mn/CeSiOx (2:1) catalyst.
Acknowledgements
This work was supported by the Natural Science Foundation of China (21546014) and the Natural Science Foundation of Shanghai, China (14ZR1417800).References
- W. Shan, Y. Geng, X. Chen, N. Huang, F. Liu and S. Yang, Catal. Sci. Technol., 2016, 6, 1195–1200 CAS
.
- S. M. Palash, M. A. Kalam, H. H. Masjuki, M. I. Arbab, B. M. Masum and A. Sanjid, Energy Convers. Manage., 2014, 77, 577–585 CrossRef CAS
- R. Guo, J. Hao, W. Pan and Y. Yu, Sep. Sci. Technol., 2015, 50, 310–321 CrossRef CAS
- G. Busca, L. Lietti, G. Ramis and F. Berti, Appl. Catal., B, 1998, 18, 1–36 CrossRef CAS
- R. Q. Long and R. T. Yang, J. Catal., 1999, 188, 332–339 CrossRef CAS
- Y. Chen, Z. Zhang, L. Liu, L. Mi and X. Wang, Appl. Surf. Sci., 2016, 366, 139–147 CrossRef CAS
- B. Thirupathi and P. G. Smirniotis, J. Catal., 2012, 288, 74–83 CrossRef CAS
- R. Guo, W. Zhen, W. Pan, J. Hong, W. Zhen, Q. Jin, C. Ding and S. Guo, J. Ind. Eng. Chem., 2013, 19, 2022–2025 CrossRef CAS
- S. S. R. Putluru, A. D. Jensen, A. Riisager and R. Fehrmann, Catal. Sci. Technol., 2011, 1, 631–637 CAS
- F. Castellino, A. D. Jensen, J. E. Johnsson and R. Fehrmann, Appl. Catal., B, 2009, 86, 196–205 CrossRef CAS
- M. Wallin, S. Forser, P. Thormählen and M. Skoglundh, Ind. Eng. Chem. Res., 2004, 43, 7723–7731 CrossRef CAS
- C. Fang, D. Zhang, S. Cai, L. Zhang, L. Huang, H. Li, P. Maitarad, L. Shi, R. Gao and J. Zhang, Nanoscale, 2013, 5, 9199–9207 RSC
- Z. Wu, B. Jiang, Y. Liu, W. Zhao and B. Guan, Appl. Catal., B, 2007, 145, 488–494 CAS
- Y. J. Kim, H. J. Kwon, I. Nam, J. W. Choung, J. K. Kil, H. Kim, M. Cha and G. K. Yeo, Catal. Today, 2010, 151, 244–250 CrossRef CAS
- P. G. Smirniotis, P. M. Sreekanth, D. A. Peña and R. G. Jenkins, Ind. Eng. Chem. Res., 2006, 45, 6436–6443 CrossRef CAS
- L. Wang, B. Huang, Y. Su, G. Zhou, K. Wang, H. Luo and D. Ye, Chem. Eng. J., 2012, 192, 232–241 CrossRef CAS
- L. Zhang, X. Zhang, S. Lv, X. Wu and P. Wang, RSC. Adv., 2015, 5, 82952–82959 RSC
- R. Jin, Y. Liu, Y. Wang, W. Cen, Z. Wu, H. Wang and X. Weng, Appl. Catal., B, 2014, 148–149, 582–588 CrossRef CAS
- J. Li, Y. Zhu, R. Ke and J. Hao, Appl. Catal., B, 2008, 80, 202–213 CrossRef CAS
- S. S. R. Putluru, A. Riisager and R. Fehrmann, Catal. Lett., 2009, 133, 370–375 CrossRef CAS
- F. Baudin, P. D. Costa, C. Thomas, S. Calvo, Y. Lendresse, S. Schneide, F. Delacroix, G. Plassat and G. Djéga-Mariadassoua, Top. Catal., 2004, 30, 97–101 CrossRef
- R. Gao, D. Zhang, P. Maitarad, L. Shi, T. Rungrotmongkol, H. Li, J. Zhang and W. Cao, J. Phys. Chem. C, 2013, 117, 10502–10511 CAS
- C. Liu, L. Chen, J. Li, L. Ma, H. Arandiyan, Y. Du, J. Xu and J. Hao, Environ. Sci. Technol., 2012, 46, 6182–6189 CrossRef CAS PubMed
- Z. Lian, F. Liu and H. He, Ind. Eng. Chem. Res., 2014, 53, 19506–19511 CrossRef CAS
- Q. Chen, R. Guo, Q. Wang, W. Pan, W. Wang, N. Yang, C. Lu and S. Wang, Fuel, 2016, 179, 852–858 CrossRef
- D. Meng, W. Zhan, Y. Guo, Y. Guo, L. Wang and G. Lu, ACS Catal., 2015, 5, 5973–5983 CrossRef CAS
- N. Yang, R. Guo, Q. Wang, W. Pan, Q. Chen, C. Lu and S. Wang, RSC Adv., 2016, 6, 11226–11232 RSC
- D. Y. Yoon, E. Lim, Y. J. Kim, J. H. Kim, T. Ryu, S. Lee, B. K. Cho, I. Nam, J. W. Choung and S. Yoo, J. Catal., 2014, 319, 182–193 CrossRef CAS
- R. Guo, Q. Chen, H. Ding, Q. Wang, W. Pan, N. Yang and C. Lu, Catal. Commun., 2015, 69, 165–169 CrossRef CAS
- K. Wijayanti, K. Leistner, S. Chand, A. Kumar, K. Kamasamudram, N. W. Currier, A. Yezerets and L. Olsson, Catal. Sci. Technol., 2016, 6, 2565–2579 CAS
- J. Luo, D. Wang, A. Kumar, J. Li, K. Kamasamudram, N. Currier and A. Yezerets, Catal. Today, 2016, 267, 3–9 CrossRef CAS
- D. W. Kwon, K. H. Park and S. C. Hong, Chem. Eng. J., 2016, 284, 315–324 CrossRef CAS
- M. Magnusson, E. Fridell and H. H. Ingelsten, Appl. Catal., B, 2012, 111–112, 20–26 CrossRef CAS
- F. Liu, K. Asakura, H. He, W. Shan, S. Shi and C. Zhang, Appl. Catal., B, 2011, 103, 369–377 CrossRef CAS
- H. Wang, X. Chen, S. Gao, Z. Wu, Y. Liu and X. Weng, Catal. Sci. Technol., 2013, 3, 715–722 CAS
- X. Yao, F. Gao, Q. Yu, L. Qi, C. Tang, L. Dong and Y. Chen, Catal. Sci. Technol., 2013, 3, 1355–1366 CAS
- L. J. Liu, Z. J. Yao, Y. Deng, F. Gao, B. Liu and L. Dong, ChemCatChem, 2011, 3, 978–989 CrossRef CAS
- X. Q. Wang, J. A. Rodriguez, J. C. Hanson, D. Gamarra, A. Martínez-Arias and M. Fernández-García, J. Phys. Chem. B, 2005, 109, 19595–19603 CrossRef CAS PubMed
- R. Guo, Q. Wang, W. Pan, Q. Chen, H. Ding, X. Yin, N. Yang, C. Lu, S. Wang and Y. Yuan, J. Mol. Catal. A: Chem., 2015, 407, 1–7 CrossRef CAS
- F. Wang, H. Dai, J. Deng, G. Bai, K. Ji and Y. Liu, Environ. Sci. Technol., 2012, 46, 4034–4041 CrossRef CAS PubMed
- L. Zhang, D. Zhang, J. Zhang, S. Cai, C. Fang, L. Huang, H. Li, R. Gao and L. Shi, Nanoscale, 2013, 5, 9821–9829 RSC
- B. Q. Jiang, Y. Liu and Z. B. Wu, J. Hazard. Mater., 2009, 162, 1249–1254 CrossRef CAS PubMed
- F. Liu, H. He, Y. Ding and C. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS
- R. Guo, W. Zhen, W. Pan, Y. Zhou, J. Hong, H. Xu, Q. Jin, C. Ding and S. Guo, J. Ind. Eng. Chem., 2014, 20, 1577–1580 CrossRef CAS
- S. Watanabe, X. Ma and C. Song, J. Phys. Chem. C, 2009, 113, 14249–14257 CAS
- R. Guo, C. Lu, W. Pan, W. Zhen, Q. Wang, Q. Chen, H. Ding and N. Yang, Catal. Commun., 2015, 59, 136–139 CrossRef CAS
- H. He, H. X. Dai and C. T. Au, Catal. Today, 2004, 90, 245–254 CrossRef CAS
- C. Hu, Y. Z. Wang and H. X. Tang, Appl. Catal., B, 2001, 30, 277–285 CrossRef CAS
- N. Y. Topsoe, J. A. Dumesic and H. Topsoe, J. Catal., 1995, 151, 241–252 CrossRef CAS
- P. Wang, Q. Wang, X. Ma, R. Guo and W. Pan, Catal. Commun., 2015, 71, 84–87 CrossRef CAS
- J. C. Dupin, D. Gonbeau, P. Vinatier and A. Levasseur, Phys. Chem. Chem. Phys., 2000, 2, 1319–1324 RSC
- N. Yang, R. Guo, W. Pan, Q. Chen, Q. Wang and C. Lu, Fuel, 2016, 169, 87–92 CrossRef CAS
- W. Pan, Y. Zhou, R. Guo, W. Zhen, J. Hong, H. Xu, Q. Jin, C. Ding and S. Guo, Environ. Prog. Sustainable Energy, 2014, 33, 358–389 Search PubMed
- X. Wang, X. Li, Q. Zhao, W. Sun, M. Tade and S. Liu, Chem. Eng. J., 2016, 288, 216–222 CrossRef CAS
- L. Chen, Z. Si, X. Wu and D. Weng, ACS Appl. Mater. Interfaces, 2014, 6, 8134–8145 CAS
- Q. Zhang, C. Qiu, H. Xu, T. Lin, Z. Lin, M. Gong and Y. Chen, Catal. Today, 2011, 175, 171–176 CrossRef CAS
- W. Tian, H. Yang, X. Fan and X. Zhang, J. Hazard. Mater., 2011, 188, 105–109 CrossRef CAS PubMed
- R. Guo, Q. Wang, W. Pan, W. Zhen, Q. Chen, H. Ding, N. Yang and C. Lu, Appl. Surf. Sci., 2014, 317, 111–116 CrossRef CAS
- E. N. Ndifor, T. Garcia, B. Solsona and S. h. Taylor, Appl. Catal., B, 2007, 76, 248–256 CrossRef CAS
- M. Wallin, S. Forser, P. Thormählen and M. Skoglundh, Ind. Eng. Chem. Res., 2004, 43, 7723–7731 CrossRef CAS
- N. Y. Topsøe, Science, 1994, 265, 1217–1219 Search PubMed
- D. A. Peña, B. S. Uphade and P. G. Smirniotis, J. Catal., 2004, 221, 421–431 CrossRef
- S. Roy, B. Viswanath, M. S. Hehde and G. Mardas, J. Phys. Chem. C, 2008, 112, 6002–6012 CAS
- Z. Wu, B. Jiang, Y. Liu, H. Wang and R. Jin, Environ. Sci. Technol., 2007, 41, 5812–5817 CrossRef CAS PubMed
- Y. Liu, T. Gu, X. Weng, Y. Wang, Z. Wu and H. Wang, J. Phys. Chem. C, 2012, 116, 16582–16592 CAS
- N. Yang, R. Guo, W. Pan, Q. Chen, Q. Wang, C. Lu and S. Wang, Appl. Surf. Sci., 2016, 378, 513–518 CrossRef CAS
- N. Yang, R. Guo, Y. Tian, W. Pan, Q. Chen, Q. Wang, C. Lu and S. Wang, Fuel, 2016, 179, 305–311 CrossRef CAS
- F. Kapteijn, L. Singoredjo, A. Andreini and J. A. Moulijn, Appl. Catal., B, 1994, 3, 173–189 CrossRef CAS
- M. Adamowska, A. Krztoń, M. Najbar, P. D. Costa and G. Djéga-Mariadassou, Catal. Today, 2008, 137, 288–291 CrossRef CAS
- Q. Chen, R. Guo, Q. Wang, W. Pan, N. Yang, C. Lu and S. Wang, J. Taiwan Inst. Chem. Eng., 2016, 64, 116–123 CrossRef CAS
- Z. Lian, F. Liu and H. He, Ind. Eng. Chem. Res., 2014, 53, 19506–19511 CrossRef CAS
- Y. Chen, J. Wang, Z. Yan, L. Liu, Z. Zhang and X. Wang, Catal. Sci. Technol., 2015, 5, 2251–2259 CAS
| This journal is © The Royal Society of Chemistry 2016 |