
Effect of type of stirring on hollow fiber liquid phase microextraction and electromembrane extraction of basic drugs: speed up extraction time and enhancement of extraction efficiency
Saeed Nojavan*,
Zeinab Tahmasebi and
Saied Saeed Hosseiny Davarani
Faculty of Chemistry, Shahid Beheshti University, G. C., 1983963113, Evin, Tehran, Iran. E-mail: s_nojavan@sbu.ac.ir; Fax: +98-21-22431662; Tel: +98-21-22431662
First published on 14th November 2016
Abstract
In microextraction procedures, the stirring of the donor solution is crucial to speed up the extraction. Two different stirring modes can be used in microextraction procedures: magnetic stirring and shaking. In this study, both hollow fiber liquid phase microextraction (HF-LPME) and electromembrane extraction (EME) procedures were considered to extract some model basic compounds under the two different mentioned stirring modes. At optimized shaking-assisted EME conditions (extraction time, 15 min; pH of acceptor solution, 2; pH of donor solution, 5; and shaking rate, 3000 rpm), 72–88% recoveries were achieved. The relative standard deviations of the analysis were found to be in the range of 6.7–13.5% (n = 5) and limits of detection ranged from 0.3 to 1.5 ng mL−1. In both of the mentioned extraction procedures, the use of a shaker was found to be associated with higher extraction rate and efficiency. The use of a shaker at the same rpm increased the area under the peak by a factor of 1.5, as compared to that of the magnetic stirrer. Using the shaker, 40% and 20% reductions were observed in optimal extraction times for EME and HF-LPME, respectively. Therefore, compared to HF-LPME method, EME was found to have larger contributions from the type of stirring. Also, results showed that the type of stirring was more important as the sample solution volume was increased (2–9 mL) while extraction efficiency was independent of stirring type for a low volume of sample solution (2 mL).
Introduction
Owing to the complexity of sample matrices and low levels (ng mL−1) of analytes, sample pretreatment and enrichment processes are crucial steps in analytical procedures.1 Hollow fiber liquid-phase microextraction (HF-LPME) is a well-known sample preparation method and has been developed extensively during the last two decades. Even though HF-LPME is a remarkably simple and effective sample preparation method, it is a relatively time-consuming technique.2 However, one can improve kinetics of HF-LPME by applying an electric potential difference over the supported liquid membrane (SLM) to establish a system termed electromembrane extraction (EME).3 Analytes are transferred into the acceptor solution via either diffusion, in HF-LPME, or a combination of diffusion and electromigration, in EME, with interfering matrix components and solid particles retained on the SLM. Several items of literature have reviewed EME4–10 and HF-LPME.11–13There are important parameters to be optimized in either of the procedures (EME and HF-LPME), including pH of the donor and acceptor solutions, choice of an organic solvent as SLM, agitation or stirring rate, and extraction time.14 Among others, agitation or stirring of the donor solution is crucial to speed up either of the extraction approaches.15–17 Although extraction could take place under no agitation in EME procedure, the chromatographic signals were significantly weaker than those recorded with agitation.18 One of the advantages of liquid-phase extraction methods based on hollow fiber is the application of high stirring rates which tend to reduce the required time for the extraction of analyte(s). Indeed, stirring rate plays an essential role in increasing the kinetics and efficiency of the extraction by increasing the mass transfer while reducing the thickness of the double layer around SLM.19 There are some reports in the literature about vortex assisted LPME (VA-LPME).20–23 The obtained results in these reports showed that vortex provided effective and mild mixing of the sample solution and increased the contact between analytes and boundary layers of the hollow fibre, thereby enhancing mass transfer rate and leading to high extraction efficiency of the target analytes. Investigations show that the magnetic stirring procedure, rather than the shaking one, has been associated with relatively longer extraction times yet lower extraction recoveries, indicating higher efficiency of the shaking procedure. Meanwhile, to the best of our knowledge, there is no report on the application of vortex shaking on electromembrane extraction and both stirring procedures to extract the same compound.
In this study, both HF-LPME and EME procedures were considered to extract some model basic compounds (imipramine, desipramine, clomipramine, trimipramine) using two different stirring modes, namely magnetic stirring and vortex shaking. For this purpose, all effective parameters on the extraction were optimized prior to investigating the effect of stirring mode.
Experimental
Reagents and materials
All model compounds (imipramine, desipramine, clomipramine, trimipramine) were obtained from Tofigh Darou Pharmaceutical Company (Tehran, Iran). HPLC grade water was obtained through a Milli-Q® system (Millipore, Milford, MA, USA) and was used to prepare all solutions. Analytical grade H3PO4, HCl and NaOH were purchased from Merck (Darmstadt, Germany). 2-Nitrophenyl octyl ether (NPOE), KH2PO4, 1-octanol, 2-ethyl hexanol, 1-heptanol, 1-hexanol were purchased from Fluka (Buchs, Switzerland).Standard solutions
Stock solution containing 1 mg mL−1 of each drug was prepared in HPLC grade water. The stock solution was protected from light exposure (using aluminum foil) and stored for a month at 4 °C. Then the required working standard solutions were freshly prepared by appropriate dilution of the stock solution typically with water, HCl or NaOH solutions (0.5, 1.0 or 2.0 M) to the required concentrations.Chromatographic conditions
The chromatographic system used was an Agilent Technologies 1200 series system consisting of a solvent degasser (G1322A), a quaternary pump (G1311A), a manual injection valve (G1328B) equipped with a 20 μL injection loop and a variable wavelength UV-Vis detector (G1314B). Separations were carried out on an Eclipse XDB-C18 HPLC column (150 × 4.6 mm, 5 μm) (Agilent Technologies, CA, USA). The data acquisition was performed by using ChemStation software (Agilent Technologies). The mobile phase consisted of A: 10 mM phosphate buffer (pH 6) and B: acetonitrile. The flow rate was set to 1.3 mL min−1. A linear gradient elution was set as (time/% mobile phase-B) 0/30, 5/30, 15/80, 25/80, 25.1/20 for the separation of selected drugs. The wavelength used for UV-Vis detector was 210 nm for all compounds.Apparatus and extraction procedure
The hollow fiber used as the support for the organic membrane and for housing the acceptor solution was a PP Q3/2 polypropylene hollow fiber with an internal diameter of 1.2 mm, wall thickness of 300 μm and pore size of 0.2 μm (Membrana, Wup-pertal, Germany). The upper end of the fiber was connected to the end of a polypropylene pipette which served as a guiding tube for the introduction of the acceptor solution. As a sample compartment, equipped with a screw cap, a 7.0 mL glass vial of a height of 6.5 cm and an internal diameter of 12 mm (unknown supplier) was used. The set-up and the equipment used in the EME experiments were identical and independent of the conditions established in the HF-LPME experiments, except for the additional electrodes and power supply. Platinum wires of a diameter of 0.25 mm were used as electrodes in the donor and acceptor solutions; they were connected to a EPS-600Z DC power supply (Paya Pajohesh Pars, Tehran, Iran) with programmable voltage in the range 0–600 V, providing currents in the range 0–0.5 A. In all experiments, magnetic stirring was done using a magnetic stirrer (Heidolph, Germany) and vortex shaking was done by a Vortex 3 shaker (IKA, Germany).Experiments were performed according to the following procedure: 6.0 mL of sample solution was filled into the sample compartment. A 31 mm-piece of the polypropylene hollow fiber was then inserted through the cap of the sample compartment and then dipped, for 15 s, into the organic solvent serving as the SLM followed by removing the excess of solvent with a medical wipe. Next, μL of the acceptor solution was introduced into the lumen of the hollow fiber via a micropipette insofar as the excess of acceptor solution was removed from the end of it. Subsequently, the lower end of the hollow fiber was sealed using a pair of hot flat-tip pliers. The fiber was then directed through the septum of the screw cap and placed into the donor solution previously filled in the sample compartment. Up to this stage, the procedure was identical for either of HF-LPME or EME. However, specifically for the EME experiments, before the extraction unit was placed on the shaker board, the negative electrode (cathode; with regard to the charge of the basic drugs (positive)) was introduced into the lumen of fiber. The fiber containing the electrode, SLM and the acceptor solution was inserted into the sample solution. The positive electrode (anode), on the other hand, was located directly into the sample solution. Subsequently, the power supply was turned on, applying the desired voltage between the two electrodes for a prescribed time. During all extractions, the sample solution was under stirring. Finally, the power supply was turned off and the hollow fiber was taken out of the donor solution. In both HF-LPME and EME procedures, the acceptor solution was collected using a 50 μL-micro-syringe and analyzed on an HPLC system.
Results and discussion
Stirring is a necessary practice to reduce the thickness of the boundary layer between the sample solution and SLM in all diffusion-based microextraction techniques.24 In EME, mass transfer is entirely performed via electrokinetic migration, diffusion and convection.25 With increasing the agitation or stirring rate, convection in the bulk solution occurs and decreases the boundary layer thickness, both of which phenomena are known to positively contribute into the extraction process. The importance of stirring rate in EME will become more significant as the sample solution volume increases, because in low-volume samples, the electric field strength is strong enough to pull the analyte through the membrane even without stirring.25 It has been seen that the extraction efficiency increases with increasing the stirring rate,24 i.e. the choice of stirring method can affect extraction efficiency and rate.Optimization of EME and HF-LPME procedures
To obtain the maximum extraction recoveries for determination of the model compounds, effective parameters on EME and HF-LPME procedures (e.g. membrane composition, applied voltage (for EME), extraction time, pH of donor and acceptor phases, stirring rate (with either of shaker or magnetic stirrer), and sample solution volume) were optimized. All optimizations were done in ultra-pure water.SLM composition
In this section, experiments were undertaken with different organic solvents serving as artificial liquid membrane. There are specific requirements for a solvent to be used as a SLM in either HF-LPME or EME methods.26–28 The SLM solvent tends to affect diffusion coefficient of analytes and determines the magnitude of applied voltage. The tendency of the analytes to distribute into the SLM should be higher than that into aquatic media to allow mass transfer of the analytes into the acceptor phase via the SLM. Furthermore, the solvent should be immiscible with water to avoid losses through the hollow fiber membrane wall pores and dissolution in the sample during the extraction. These criteria are similar for HF-LPME and EME procedures. In EME procedure, it is crucial for the organic solvents to have sufficient electrical conductivity so as to allow a continuous electric field across the entire system.29 In most EME procedures NPOE was used (as SLM solvent) for the extraction of basic compounds and alcohols such as 1-octanol and 2-ethyl hexanol were used for extraction of different classes of compounds in HF-LPME procedure.30Considering all of the above criteria, alcohols such as 1-hexanol, 1-heptanol, 1-octanol, NPOE (just for EME) and 2-ethyl hexanol were evaluated. Based on the evaluation results, NPOE was found to be an efficient organic solvent for electrokinetic migration of basic drugs through the SLM, while 2-ethyl hexanol was indicated as an efficient organic solvent for the extraction of basic drugs in HF-LPME procedure (Fig. 1). In these evaluations, the sample solution was stirred on a shaker. The obtained results in this study are accordance with literature.29
 | ||
| Fig. 1 Effect of composition of SLM in EME and HF-LPME on the extraction efficiencies. Other extraction conditions: pH of sample solution: 3 (A) and 11 (B), pH of acceptor solution: 2 (A and B), extraction time: 5 min (A) and 20 min (B), shaking speed: 1500 rpm (A and B), extraction voltage: 50 V (A). Error bars were obtained based on 3 replicates. | ||
Applied voltage in EME
The main mass transfer mechanism in EME is the electrokinetic migration of the analytes across the SLM into the acceptor solution. Thus, the applied voltage across a SLM is an important factor for efficient extraction of basic drugs in EME.26 In this study, looking for an optimal applied voltage, a series of experiments were conducted with extraction voltages ranging from 0 to 300 V. Summarized in Fig. 2, the results showed that the extraction efficiency increased with increasing the voltage from 0 to 300 V. In other researches it was shown that when NPOE was used as the SLM solvent higher voltages could be used.31 However, reproducibility of the results decreased at higher voltages, so that voltages above 300 V led, even though occasionally, to loss of acceptor phase. Thus, the voltage of 300 V was selected as the optimal voltage for the rest of the study. | ||
| Fig. 2 Effect of applied voltage on the extraction efficiencies. Other extraction conditions: pH of sample solution: 3.0, pH of acceptor solution: 2.0, extraction time: 5 min, shaking speed: 1500 rpm, SLM solvent: NPOE. Error bars were obtained based on 3 replicates. | ||
Effect of pH of donor and acceptor solutions
In EME procedure, the analyte should be in its ionic form. Also, it was shown that the ratio of total ionic concentration of the donor phase to that of the acceptor phase, which is referred to as ion balance (χ), affects the flux over the membrane, so that the flux may be declined with an increase in ion balance, as described by theoretical models.32,33 In order to investigate the influence of pH of the acceptor and donor phases, pH of the acceptor phase was kept constant at 2.0 while varying the pH of the donor phase within the range of 2–10. Results showed that, maximum extraction recoveries were obtained when pH of the donor phase was adjusted to 5.0. By increasing the pH in the acceptor solution, ion balance (χ) decreased, accelerating the flux of the ions into the acceptor phase.30 Accordingly, pH of the donor phase was kept constant at 5.0 while varying pH of the acceptor phase in the range of 2–4. Here, the maximum extraction recoveries were obtained at the acceptor phase pH of 2.0. Thus, further experiments were done with pH values of the acceptor and donor phases adjusted to 2.0 and 5.0, respectively (Fig. 3A and B).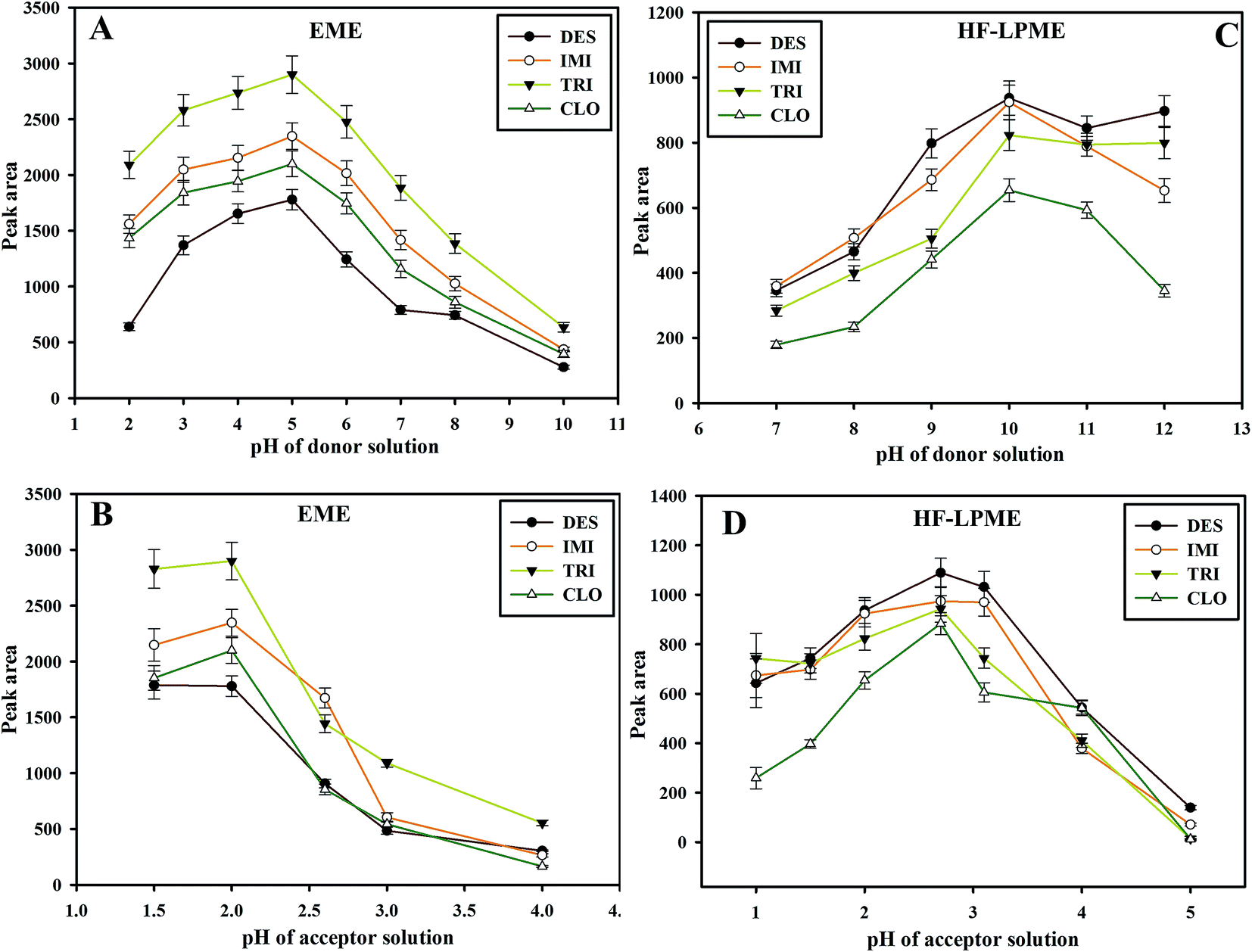 | ||
| Fig. 3 Effect of (A) pH of donor solution in EME, (B) pH of acceptor solution in EME, (C) pH of donor solution in HF-LPME and (D) pH of acceptor solution in HF-LPME on the extraction efficiencies. Other extraction conditions: pH of acceptor solution: 2.0 (A and C), pH of donor solution: 5 (A) and 10 (C), extraction time: 5 min (A and B) and 20 min (C and D), shaking speed: 1500 rpm (A–D), extraction voltage: 300 V (A and B), SLM solvent: NPOE (A and B) and 2-ethyl hexanol (C and D). Error bars were obtained based on 3 replicates. | ||
In HF-LPME procedure, for basic analytes, donor phase should be alkalized to ensure their deprotonation and, consequently, reduce their solubility in sample solution. The acceptor phase should be acidic to provide high solubility for the basic analytes and ionize them to prevent re-entering of the analytes into the organic phase. In a series of experiments, pH of the donor phase was varied from 7.0 to 12.0. Best peak areas were observed at pH 10. The effect of the acceptor phase pH on extraction recovery was studied at the range of 1.0–5.0. The results (Fig. 3C and D) showed the maximum extraction recovery at pH 2.7.
Effect of stirring type and rate
Following with the research, the effect of stirring type and rate was studied on both of the EME and HF-LPME methods. For this purpose, first, the stirring rate was examined using the shaker applied at 0–3000 rpm. The results showed that an increase in stirring rate increases the efficiency of the extraction. Indeed, higher stirring rates tend to develop full circulation of analytes, increasing the chance of being the species to be within SLM region, leading to the elimination of the concentration gradient in donor phase and probably in acceptor phase. The use of shaker induces vibrations into the donor phase and probably in SLM and acceptor phase, finally resulting in the elimination of the concentration gradients in both phases. On the other hand, the vibration created by the shaker reduces the thickness of the boundary layer in the SLM, accelerating the entrance of the analytes into the acceptor phase. Therefore, the stirring rate of 3000 rpm was chosen as the optimal stirring rate. It is worth noting that as the maximum rpm at which the used shaker in this research was capable of operating was just 3000 rpm, we left with no chance to test any higher stirring rate. The effect of shaking rate is demonstrated in Fig. 4A. In the following, in order to compare the shaking mode to the magnetic stirring mode, a magnetic stirrer was used for stirring, using which the stirring was applied at various rates within the range of 250 to 1400 rpm. As shown in Fig. 4B, the extraction efficiency of pharmaceutical compounds was observed to increase with stirring rate up to 1400 rpm. However, since the maximum affordable rate of the magnetic stirrer was 1400 rpm, we left with no chance to test any higher rate. But as expected, the corresponding area under the extraction peak to magnetic stirrer was approximately one third of that of the case where the shaker was applied. | ||
| Fig. 4 Effect of (A) shaking speed in EME, (B) stirring speed in EME, (C) shaking speed in HF-LPME and (D) shaking speed in HF-LPME on the extraction efficiencies. Other extraction conditions: pH of acceptor solution: 2.0 (A and B) and 2.7 (C and D), pH of donor solution: 5 (A and B) and 10 (C and D), extraction time: 5 min (A and B) and 20 min (C and D), extraction voltage: 300 V (A and B), SLM solvent: NPOE (A and B) and 2-ethyl hexanol (C and D). Error bars were obtained based on 3 replicates. | ||
Similar comparison was performed between the shaker and the magnetic stirrer method but in HF-LPME procedure. For this purpose, first, the stirring rate was varied within 0–3000 rpm using the shaker. In this way, with increasing the stirring rate to up to 3000 rpm, the extraction efficiency of basic pharmaceutical compounds was observed to be increased (Fig. 4C). However, the method had its reproducibility decreased with further increase in the stirring rate, due to bubbles creation in the solution, membrane thickness changing and solvent leakage into the acceptor phase.34 Subsequently, the magnetic stirrer was used at stirring rates within the range of 250–1400 rpm. As shown in Fig. 4D, the extraction efficiency of the pharmaceutical compounds increased with stirring rate to up to 1400 rpm. Therefore, at the same rpm, the shaker was associated with about 1.5 times as large increase in the area under the peak extraction as with the magnetic stirrer in either of EME or HF-LPME procedures.
Also, Fig. 4 shows that extraction efficiencies of both EME and HF-LPME procedures increased with increasing shaking and magnetic stirring rates exponentially except for Fig. 4B. This comparison indicates that ions accumulation on both sides of SLM (boundary layer) in EME procedure can be reduced by shaking more than magnetic stirring. While there is no ions accumulation in HF-LPME procedure.
Effect of sample volume
The sample volume affects the efficiency of the convection; thus, it influences the extraction efficiency. In other hand, the sample volume and the stirring rate are related variables. Overall, when higher samples volumes are processed, higher stirring rates can be used. In this experiment, for investigation of stirring behavior, the developed EME and HF-LPME were performed on four different volumes of the sample (2, 4, 6 and 9 mL) containing same amounts of analytes. Also, the effect of type of stirring in different volumes was investigated. The both stirring modes were applied in similar rates (1400 rpm) and other extraction conditions were constant. The results showed that areas under peaks obtained by both EME and HF-LPME procedure decreased by decreasing the sample volume (Fig. 5A–D). Also, results indicated similar efficiency for both magnetic stirrer and shaker when low volume (2 mL) of samples were used while significant difference in extraction efficiency was obtained between shaking and magnetic stirrer when high volume (9 mL) of samples were used. Therefore, at the same rpm (1400 rpm) and high volume of sample (9 mL), the shaker was associated with about 2 times as large increase in the area under the peak extraction as with the magnetic stirrer in either of EME or HF-LPME procedures.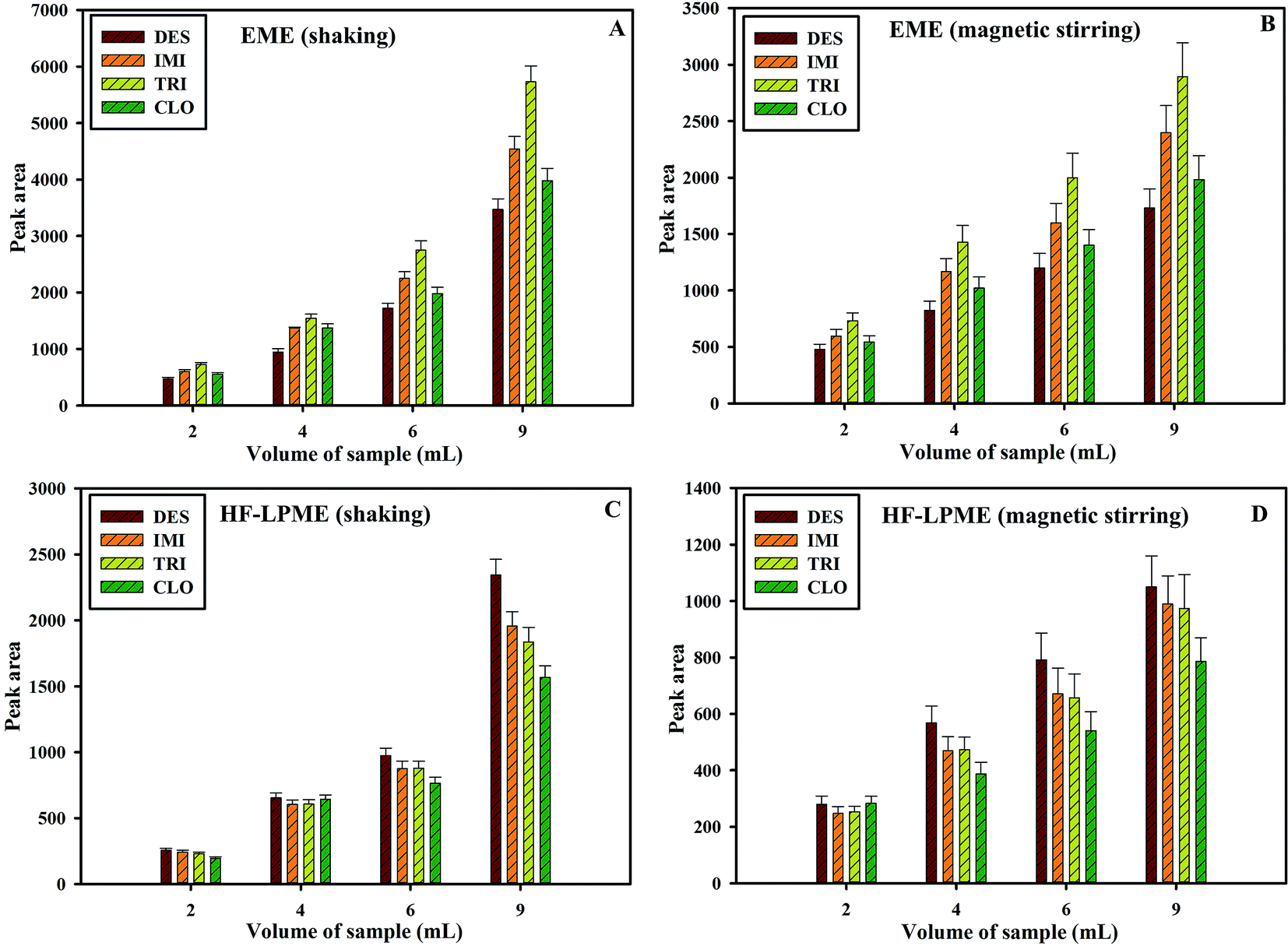 | ||
| Fig. 5 Effect of sample volume in (A) shaking-assisted EME, (B) magnetic stirrer-assisted EME, (C) shaking-assisted HF-LPME and (D) magnetic stirrer-assisted HF-LPME on the extraction efficiencies. Other extraction conditions: pH of acceptor solution: 2.0 (A and B) and 2.7 (C and D), pH of donor solution: 5 (A and B) and 10 (C and D), shaking speed: 1400 rpm (A and C), magnetic stirring speed: 1400 rpm (B and D), voltage: 300 V (A and B), SLM solvent: NPOE (A and B) and 2-ethyl hexanol (C and D). Error bars were obtained based on 3 replicates. | ||
Effect of type of stirring on extraction time
In this section, the effect of type of stirring on extraction time and, consequently, extraction efficiency is investigated. Using a shaker, stirring was applied at 3000 rpm, with the extraction time varied from 5 to 30 minutes (for EME procedure). The results showed that the extraction efficiency increased with increasing the extraction time to up to 15 minutes, after which time the extraction efficiency decreased (Fig. 6A). Such a behavior could be explained by two reasons. First, as time passes, organic solvent content of the membrane is lost, reducing the extraction efficiency. Second, over time, there are chances that a double layer of ions forms across the membrane; the layer can provide a kind of resistance against analyte mass transfer, again the extraction efficiency consistency.35 Finally, 15 min was selected as the optimum time for EME procedure. | ||
| Fig. 6 Effect of extraction time in (A) shaking-assisted EME, (B) magnetic stirrer-assisted EME, (C) shaking-assisted HF-LPME and (D) magnetic stirrer-assisted HF-LPME on the extraction efficiencies. Other extraction conditions: pH of acceptor solution: 2.0 (A and B) and 2.7 (C and D), pH of donor solution: 5 (A and B) and 10 (C and D), shaking speed: 3000 rpm (A and C), stirring speed: 1400 rpm (B and D), voltage: 300 V (A and B), SLM solvent: NPOE (A and B) and 2-ethyl hexanol (C and D). Error bars were obtained based on 3 replicates. | ||
In order to compare the corresponding optimal extraction times to the shaker and magnetic stirrer, experiments were conducted using the magnetic stirrer where stirring rate was set at 1400 rpm (optimal value) with the extraction time being varied from 5 to 30 min. The results showed that the extraction efficiency increased with increasing the extraction time to up to 25 min, after which time the extraction efficiency exhibited a reduction (Fig. 6B). Therefore, compared to the magnetic stirrer, the shaker was associated with 40% shorter extraction time.
Further experiments were conducted to obtain associated optimal extraction times with the stirring methods when applied in HF-LPME. For this purpose, first, a shaker was used with its stirring rate set to 3000 rpm. Extraction time was varied from 10 to 50 min. The results showed that the extraction efficiency increased with increasing time to up to 40 min, after which time the efficiency decreased (Fig. 6C). The behavior could be attributed to three facts: (1) mass transfer process is time-dependent, (2) the extractable amount of analyte depends on the analyte partition coefficient between the aqueous solution and extraction solution, and (3) HF-LPME is an equilibrium process and longer extraction times tend to causes a sort of resistance against mass transfer.36
Then in order to compare associated optimal extraction times with shaker and magnetic stirrer, the magnetic stirrer was used for stirring at 1400 rpm. Accordingly, the extraction time was varied from 10 to 60 min. The results showed that the extraction efficiency increased with increasing the extraction time to up to 50 min, after when the extraction efficiency decreased (Fig. 6D).
So, as expected, even though in both of the EME and HF-LPME methods, the use of shaker was found to be associated with shorter optimal extraction times, but the reductions were measured at 40% and 20% in EME and HF-LPME methods, respectively. Therefore, type of stirring was found to be of larger contribution into EME rather than HF-LPME; it can be a result of the fact that, mass transfer in EME goes through diffusion, migration, and convection,25 while in LPME, it proceeds via diffusion and convection only.35
Validation of method
Because EME method provided higher efficiency in a shorter time, it was selected as the ultimate method to analyze the model compounds. Linear range of the method was tested at 8 different concentration levels; based on the results, the linear range was calculated to be 1–1000 ng mL−1, with the corresponding correlation coefficients being within 0.995–0.999. Repeatability was determined by performing five analyses at 50 ng mL−1 for each analyte, based on which results the relative standard deviation was obtained to range within 6.7–13.5%. Enrichment factor was calculated to range within 144–176, corresponding to extraction efficiencies in the range of 72–88%. Therefore, calculated based on signal-to-noise ratio, limits of detection (LOD) was found to fall within 0.3–1.5 ng mL−1. All results are detailed in Table 1.Conclusions
Comparison of obtained results by shaking-assisted and magnetic stirrer-assisted extraction procedures (in both of EME and HF-LPME) showed that shaking provided effective and mild mixing of the sample solution and increased the contact between analytes and boundary layers of the hollow fibre, thereby enhancing mass transfer rate and leading to high extraction efficiency of the target analytes and short extraction times. This may be a result of the vibrations created by shaker which not only contribute to the stirring of the donor phase, but also induce a kind of stirring into the SLM and acceptor phase, so that the gradient of concentration disappears in both donor and acceptor phases; also, the shaker reduces the thickness of the double layer surrounding hollow fiber, accelerating the introduction of the analytes into the acceptor phase. The use of the shaker increased areas under the peaks by 1.5–2.0 times as large as those obtained by the magnetic stirrer, and reduced the extraction times by up to 10 min in both of EME and HF-LPME. Also investigation of sample volume indicated large difference between two different stirring modes when sample volume was increased. Further, type of stirring was found to impose larger effects on EME rather than HF-LPME.Conflict of interest
There are no financial or commercial conflicts of interest.Abbreviations
| EME | Electromembrane extraction |
| HF-LPME | Hollow fiber liquid phase microextraction |
| NPOE | 2-Nitrophenyl octyl ether |
| SLM | Supported liquid membrane |
Acknowledgements
Financial support from the Research Affairs of Shahid Beheshti University is gratefully acknowledged.References
- S. Nojavan, Z. Tahmasebi, T. Bidarmanesh, H. Behdad, M. Nasiri-Aghdam, S. Mansori and A. Pourahadi, J. Sep. Sci., 2013, 36, 3256–3263 CAS
.
- T. G. Halvorsen, S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. B, 2001, 760, 219–226 CrossRef
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2006, 1109, 183–190 CrossRef CAS PubMed
- A. Wuethrich, P. R. Haddad and J. P. Quirino, Trends Anal. Chem., 2016, 80, 604–611 CrossRef CAS
- K. F. Seip, A. Gjelstad and S. Pedersen-Bjergaard, Bioanalysis, 2015, 7, 463–480 CrossRef CAS PubMed
- A. Gjelstad and S. Pedersen-Bjergaard, Anal. Methods, 2013, 5, 4549–4557 RSC
- S. Pedersen-Bjergaard and K. E. Rasmussen, Trends Anal. Chem., 2008, 27, 934–941 CrossRef CAS
- G. Morales-Cid, S. Cardenas, B. M. Simonet and M. Valcarcel, Trends Anal. Chem., 2010, 29, 158–165 CrossRef CAS
- P. Kuban, A. Slampova and P. Bocek, Electrophoresis, 2010, 31, 768–785 CrossRef CAS PubMed
- Y. Yamini, S. Seidi and M. Rezazadeh, Anal. Chim. Acta, 2014, 814, 1–22 CrossRef CAS PubMed
- M. A. Bello-Lopez, M. Ramos-Payan, J. A. Ocana-Gonzalez, R. Fernandez-Torres and M. Callejon-Mochon, Anal. Lett., 2012, 45, 804–830 CrossRef CAS
- J. Lee, H. K. Lee, K. E. Rasmussen and S. Pedersen-Bjergaard, Anal. Chim. Acta, 2008, 624, 253–268 CrossRef CAS PubMed
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2008, 1184, 132–142 CrossRef CAS PubMed
- M. Balchen, H. Jensen, L. Reubsaet and S. Pedersen-Bjergaard, J. Sep. Sci., 2010, 33, 1665–1672 CrossRef CAS PubMed
- M. Balchen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1152, 220–225 CrossRef CAS PubMed
- S. Pedersen-Bjergaard and K. E. Rasmussen, J. Chromatogr. A, 2006, 1109, 183–190 CrossRef CAS PubMed
- A. Gjelstad, T. M. Andersen, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2007, 1157, 38–45 CrossRef CAS PubMed
- L. Xu, P. C. Hauser and H. K. Lee, J. Chromatogr. A, 2008, 1214, 17–22 CrossRef CAS PubMed
- S. Seidi, Y. Yamini, A. Heydari, M. Moradia, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS PubMed
- M. Asadi, A. M. Haji Shabani and S. Dadfarnia, J. Sep. Sci., 2016, 39, 2238–2245 CrossRef CAS PubMed
- P. Wang, Y. Xiao, W. Liu, J. Wang and Y. Yang, Food Chem., 2015, 172, 385–390 CrossRef CAS PubMed
- Y. Li, G. Yang, J. Zhao and Y. Yang, J. Braz. Chem. Soc., 2014, 25, 1512–1519 CAS
- C. Bosch Ojeda and F. S. Anchez Rojas, Chromatographia, 2014, 77, 745–754 CAS
- J. Lee, F. Khalilian, H. Bagheri and H. K. Lee, J. Chromatogr. A, 2009, 1216, 7687–7693 CrossRef CAS PubMed
- S. Seidi, Y. Yamini, A. Saleh and M. Moradi, J. Sep. Sci., 2011, 34, 585–593 CrossRef CAS PubMed
- S. Seidi, Y. Yamini, A. Heydari, M. Moradi, A. Esrafili and M. Rezazadeh, Anal. Chim. Acta, 2011, 701, 181–188 CrossRef CAS PubMed
- S. Asadi and S. Nojavan, Anal. Chim. Acta, 2016, 923, 24–32 CrossRef CAS PubMed
- P. Zahedi, S. S. H. Davarani, H. R. Moazami and S. Nojavan, J. Pharm. Biomed. Anal., 2016, 117, 485–491 CrossRef CAS PubMed
- S. S. H. Davarani, A. M. Najarian, S. Nojavan and M. A. Tabatabaei, Anal. Chim. Acta, 2012, 725, 51–56 CrossRef CAS PubMed
- T. Rahmani, A. Rahimi and S. Nojavan, Anal. Chim. Acta, 2016, 903, 81–90 CrossRef CAS PubMed
- S. Nojavan, A. Pourahadi, S. S. H. Davarani, A. M. Najarian and M. Beigzadeh-Abbassi, Anal. Chim. Acta, 2012, 745, 45–52 CrossRef CAS PubMed
- M. Rezazadeh, Y. Yamini, S. Seidi and A. Esrafili, Anal. Chim. Acta, 2013, 773, 52–59 CrossRef CAS PubMed
- T. M. Middelthon-Bruer, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Sep. Sci., 2008, 31, 753–759 CrossRef CAS PubMed
- L. Fotouhi, Y. Yamini, S. Molaei and S. Seidi, J. Chromatogr. A, 2011, 1218, 8581–8586 CrossRef CAS PubMed
- I. J. O. Kjelsen, A. Gjelstad, K. E. Rasmussen and S. Pedersen-Bjergaard, J. Chromatogr. A, 2008, 1180, 1–9 CrossRef CAS PubMed
- Y. Zhang and H. K. L. Lee, J. Chromatogr. A, 2013, 1273, 12–17 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2016 |