
Palladium-catalyzed carbonylation of aryl halides: an efficient, heterogeneous and phosphine-free catalytic system for aminocarbonylation and alkoxycarbonylation employing Mo(CO)6 as a solid carbon monoxide source†
Abdol-Reza Hajipour*ab,
Zeinab Tavangar-Rizia and
Nasser Iranpoorc
aPharmaceutical Research Laboratory, Department of Chemistry, Isfahan University of Technology, Isfahan 84156, Islamic Republic of Iran. E-mail: haji@cc.iut.ac.ir; Fax: +98 311 391 2350; Tel: +98 311 391 3262
bDepartment of Neuroscience, University of Wisconsin, Medical School, 1300 University Avenue, Madison, 53706-1532 WI, USA
cDepartment of Chemistry, College of Sciences, Shiraz University, Shiraz 71454, Iran
First published on 4th August 2016
Abstract
Immobilized palladium metal-containing magnetic nanoparticles (ImmPd(0)-MNPs) were synthesized and characterized as an immobilized, phosphine-free catalyst for carbonylation reactions, namely the alkoxycarbonylation and aminocarbonylation reactions. Various substituted aryl iodides tolerated the reaction conditions and a wide variety of alcohols and amines were used efficiently. The effects of the solvent, base, and temperature were studied in both the mentioned reactions. The developed catalytic system avoids the use of phosphine ligands and can be reused for up to eight consecutive cycles. The recycled catalyst was characterized by TEM and ICP analysis.
Introduction
The transition-metal-catalyzed carbonylation reaction of aryl halides in the presence of an appropriate nucleophile represents a powerful strategy for the selective introduction of important frameworks in natural products, pharmaceuticals, agrochemicals, and functional materials.1,2 During the last few decades, considerable efforts have been performed to develop transition-metal-catalyzed carbonylation reactions for the conversion of organic halides3–5 or organic pseudohalides6,7 to carboxylic acid derivatives. Recently, Iranpoor et al. reported the nickel-catalyzed synthesis of thioesters, esters, and amides from aryl iodides in the presence of chromium hexacarbonyl.8 Among the different types of carbonylation reactions, palladium-catalyzed carbonylations are reported more frequently due to the exceptional power of palladium catalysts in carbonylative couplings compared to the other metals.The palladium-catalyzed carbonylation of aryl halides with alcohols and amines is accepted as an interesting and important chemical transformation for the synthesis of aromatic esters and amides,9 which are important building blocks for various pharmaceuticals and agrochemicals.10 Conventionally, aromatic esters have been synthesized via reaction of the carboxylic acid with alcohols.11 The carbonylation of aryl halides in the presence of an alcohol is an attractive possible method that allows use of a wide range of substrates, thus representing a great advantage for the synthesis of substituted aromatic esters and its derivatives.12–14 In this respect, various palladium-based catalytic systems have been investigated for the alkoxycarbonylation reaction.15–19 Aromatic amides are also important classes of carbonyl compounds in various natural products and designed pharmaceutical molecules. Some heterocyclic amides are potential CNS (central nervous system)-active compounds.20 Aminocarbonylation is an advantageous method for the direct one-step synthesis of aromatic amides via carbonylative coupling of aryl, heteroaryl, or alkynyl halides with primary/secondary amines.
In 1974, Heck reported the first Pd-catalyzed alkoxycarbonylation and aminocarbonylation of aryl halides with carbon monoxide.21 After this report, some other Pd-catalyzed carbonylation reactions were developed to obtain carbonylative products from aryl halides in the presence of CO gas.10a,17,22 To avoid the use of carbon monoxide in these reactions, a variety of sources, such as Mo(CO)6, carbamoylstannes, and DMF, have been used.23 Recently, Iranpoor et al. reported the aminocarbonylation of aryl halides in the presence of Mo(CO)6 as the carbonyl source,24 and also DMF/POCl3 (ref. 25) and DMF/WCl6 (ref. 26) as a new combinatorial carbonylation reagent system. Accordingly, performing carbonylation reactions in which cheaper catalysts and CO substituents can be used is of great interest.
Literature reports reveal that a variety of palladium-based homogeneous catalytic systems have been used for the alkoxycarbonylation and aminocarbonylation of aryl halides.17,27 Although the homogeneous palladium catalysts offer high selectivity and yields, and a high reaction rate and turnover numbers (TON), the main drawback of homogeneous catalysis is associated with the lack of recovery and recycling of the catalyst. Here, one solution is to use an excess of N/P-containing ligand to avoid catalyst deactivation, but this restricts the applicability of the carbonylation procedure since its separation from the reaction products and restoration is usually difficult.
To avoid these serious issues, heterogeneous Pd catalysis is a promising option. Considering this, several groups have developed the carbonylation of aryl halides by using immobilized palladium complexes on various supports, such as activated carbon,28 silica,10a,29 MCM-41,30 organic polymer,22c,31 SBA-15,32 ZIF-8,22b Fe3O4,22d and MOF-5.33 Recently, Bhanage et al. demonstrated silica-supported palladium–phosphine as a reusable catalyst for the alkoxycarbonylation and aminocarbonylation of aryl and heteroaryl iodides.34
In the current study, we wish to report the synthesis of palladium nanoparticles immobilized on magnetic methionine-functionalized chitosan (ImmPd(0)-MNPs) as a new and highly efficient heterogeneous catalyst for the alkoxycarbonylation and aminocarbonylation of aryl iodides.
Results and discussions
Synthesis and characterization of the immobilized palladium metal-containing magnetic nanoparticles (ImmPd(0)-MNPs)
As shown in Scheme 1, the catalyst was prepared in three steps. First, methionine–CS was prepared by the reaction between methyl methioninate and chitosan in DMF. Then, it was treated with Fe3O4 nanoparticles prepared by mixing FeCl3·6H2O and FeSO4 according to the reported procedure,35 to achieve Fe3O4–CS–methionine as a magnetic substrate. Finally, palladium acetate was attached to this magnetic support in ethanol to give ImmPd(0)-MNPs as the final catalyst.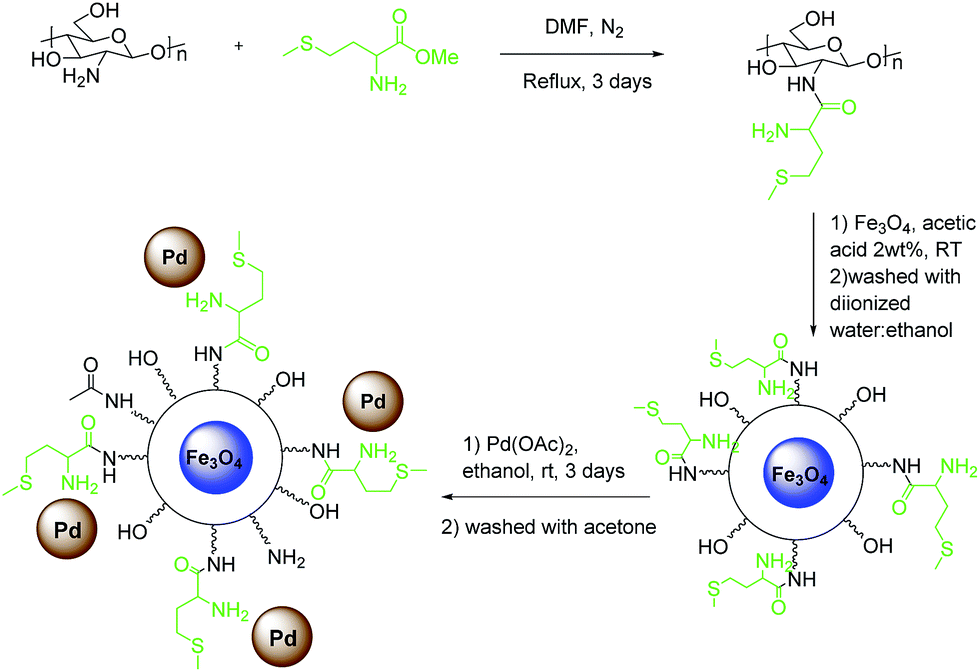 | ||
| Scheme 1 Synthesis of the Fe3O4–CS–methionine-supported Pd(0) catalyst. | ||
The prepared catalyst was characterized by transmission electron microscopy (TEM), field emission scanning electron microscopy (FE-SEM), powder X-ray diffraction (XRD), and energy dispersive X-ray spectroscopy (EDX). The attachment of Fe3O4 onto the CS–methionine support was confirmed by comparison of the FT-IR spectra (Fig. 5) of the CS–methionine before and after loading with Fe3O4. The thermal behavior of the catalyst was also investigated using a thermogravimetric analyzer (TGA) (Fig. 6). The magnetization curve of ImmPd(0)-MNPs represents the magnetic behavior of the catalyst (Fig. 7).
The TEM images of fresh and spent catalyst are shown in Fig. 1, which indicates a nearly spherical morphology of the catalyst, with an average diameter of about 6–7 nm, and a good distribution. The field emission scanning electron microscopy (FE-SEM) image in Fig. 2 illustrates the spherical external morphologies of the catalyst. Moreover, to determine the Pd content on the catalyst, the catalyst was treated with concentrated HCl to digest the Pd particles and was then analyzed by the ICP technique. The amount of palladium was detected to be 7.1 ppm (7.1 mg L−1). The energy dispersive spectroscopy analysis of the X-ray (EDAX) data for the ImmPd(0)-MNPs catalyst is given in Fig. 3.
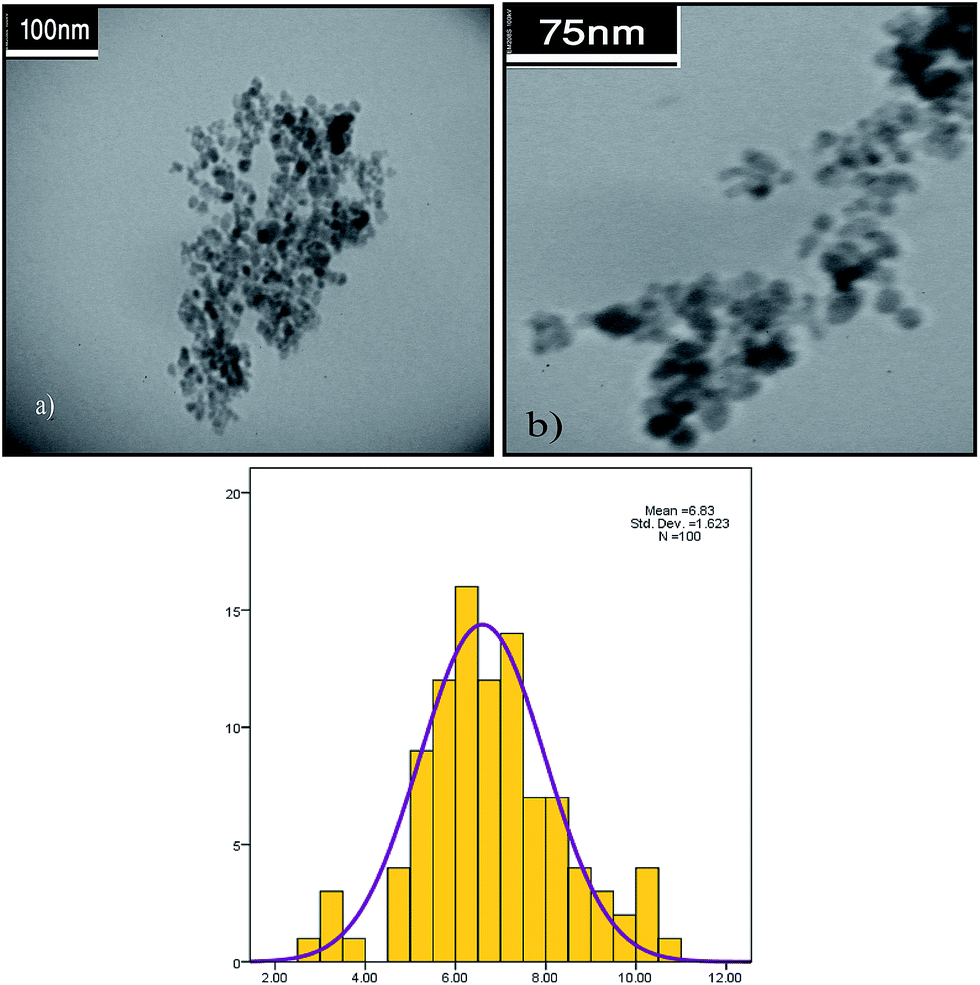 | ||
| Fig. 1 TEM image of the ImmPd(0)-MNPs catalyst, indicating the morphology of the Pd nanoparticles on the magnetic chitosan support (a). TEM image of the ImmPd(0)-MNPs catalyst after eight recycles (b). | ||
 | ||
| Fig. 2 FE-SEM images of the ImmPd(0)-MNPs catalyst. | ||
 | ||
| Fig. 3 SEM-EDX spectrum of the ImmPd(0)-MNPs catalyst. | ||
The EDAX data shows the presence of the palladium on the magnetic chitosan in the ImmPd(0)-MNPs catalyst. Moreover, the EDX spectrum specified the other elements, including S, O, and Fe, present in the Fe3O4–CS–methionine substrate. The XRD spectrum (Fig. 4) provided more evidence for the presence of palladium and iron in the catalyst. The XRD study of ImmPd(0)-MNPs illustrated the presence of highly dispersed palladium complexes in the structure of the modified-chitosan support. According to Fig. 4, a sharp peak at 2θ = 40°, which is linked to palladium, confirms the crystalline nature of the palladium species. The peaks assigned as (111), (200), (220), (311), and (222), indicate the cubic phase of the palladium nanoparticles {JCPDS 01-087-0638}. As presented in Fig. 3, 11 characteristic diffraction peaks [2θ = 30.1° (220), 35.5° (311), 43.1° (400), 47.2° (331), 53.5° (422), 57.0° (511), 62.6° (440), 75.1° (622), 82.0° (551), 86.8° (642), and 89.8° (731)] can be clearly considered for the Fe3O4 particles, which are related to the cubic phase of Fe3O4 {JCPDS 01-075-0033}.
 | ||
| Fig. 4 XRD spectrum of the ImmPd(0)-MNPs catalyst. | ||
Comparisons between the FT-IR spectra (Fig. 5) of pure chitosan, chitosan–methionine, Fe3O4, and the Fe3O4–CS–methionine substrate show that some absorption bands in Fe3O4–CS–methionine exist in both chitosan and Fe3O4. The FT-IR spectrum of CS–methionine revealed a carbonyl group absorption band, which confirmed methionine was bound into the chitosan after the reaction between chitosan and methioninate. A characteristic strong and broad band appeared at around 3419 cm−1, assigned to the stretching vibration of the –OH group, the extension vibration of the N–H group, and inter-hydrogen bonds of polysaccharide in the Fe3O4–CS–methionine spectrum.36,37 In addition, the intensity of the band at 1385 cm−1 increased in the spectrum of the CS–methionine material due to the introduction of additional amine groups.36 In the FT-IR spectrum, bands at 2925, 2354, 1632, 1394, 1064, and 566 cm−1 correspond to the bonds in Fe3O4–CS–methionine. The band at 566 cm−1 is attributed to the Fe–O stretching vibration of Fe3O4.38 An additional band at 1632 cm−1 is representative of the (–CON–) carboxyl amid group vibration of the methionine moiety.
 | ||
| Fig. 5 FT-IR spectra of pure chitosan, CS–methionine, Fe3O4, and Fe3O4–CS–methionine substrate. | ||
The TGA analysis results (Fig. 6) show that two main weight losses occurred. The first one, which happened at around 100 °C, was assigned to loss of the adsorbed solvent molecules and moisture.39 The second one, above 200 °C, was associated to the decomposition of the polysaccharide chain. So, the TGA analysis confirmed the high thermal stability of the catalyst.
 | ||
| Fig. 6 Typical TGA curve of the ImmPd(0)-MNPs catalyst. | ||
The magnetization curves of ImmPd(0)-MNPs and Fe3O4 indicated the magnetic behavior of ImmPd(0)-MNPs (Fig. 7). The two samples show zero coercivity and remanence on the magnetization curve. The lack of a hysteresis loop displays the magnetic properties and the ability of the catalyst to be separated efficiently from the reaction mixture by an external magnetic force. In addition, the decrease in the magnetization value of the catalyst in comparison with Fe3O4 shows that the surface of Fe3O4 is concealed by the attachment of chitosan molecules through their bond formation via their hydroxyl functional groups. However, even with this reduction in the saturation magnetization, the catalyst still can be separated simply from the solution by an external magnet.
 | ||
| Fig. 7 Magnetization curves of ImmPd(0)-MNPs and Fe3O4. | ||
Catalytic activities
Since supported palladium catalysts have displayed high catalytic activity in a wide range of industrial main processes and have been widely studied for C–C coupling,40 after characterizing the catalyst, we decided to explore the catalytic properties of the ImmPd(0)-MNPs in the field of carbonylation reactions (Scheme 2). | ||
| Scheme 2 Palladium-catalyzed alkoxycarbonylation and aminocarbonylation of aryl iodides. | ||
Alkoxycarbonylation of aryl iodides catalyzed by ImmPd(0)-MNPs
To test the catalytic activity of the present catalyst, we performed the reaction of iodobenzene (1a) with methanol (2a′) as a model reaction to identify the effective conditions for promoting Pd-carbonylation. The reaction was carried out under various conditions (Table 1). Initially, the reaction was carried out under solvent-free conditions in the presence of K2CO3 as the base. In this condition, the O-arylated product (4a′′) was obtained in higher yield than the carbonylated product (3aa′) (Table 1, entry 1). It was also found that by employing water as the solvent, the corresponding ester product was obtained only in an approximately 40% yield, with the C–O coupling product (37%) and benzoic acid (15%) observed as the side-products (Table 1, entry 2). Iodobenzene provided methyl benzoate as the major product, with an approximately 62% yield in DMF, with the formation of an O-arylated side-product (21%) (Table 1, entry 3).
|
|||||
|---|---|---|---|---|---|
| Entry | Solvent | Base | T/°C | 2a (%) | 3a (%) |
| a Reaction conditions: iodobenzene (0.5 mmol), methanol (0.75 mmol), base (1.5 mmol), catalyst (0.01 g, 0.14 mol%), Mo(CO)6 (0.5 mmol), solvent (4 mL), time (2 h).b Excess amount of methanol was used.c The amount of catalyst was 0.005 g (0.07 mol%).d 0.5 mmol of methanol was used. | |||||
| 1b | — | K2CO3 | 100, reflux | 27 | 70 |
| 2 | H2O | K2CO3 | 80, reflux | 43 | 37 |
| 3 | DMF | K2CO3 | 100 | 62 | 21 |
| 4 | DMF | KOH | 100 | 60 | 28 |
| 5 | DMF | K3PO4 | 100 | 57 | 33 |
| 6 | DMF | Bu3N | 100 | 90 | 6 |
| 7 | DMF | Bu3N | 80 | 90 | 8 |
| 8 | DMF | Bu3N | 60 | 42 | 8 |
| 9c | DMF | Bu3N | 80 | 58 | 4 |
| 10d | DMF | Bu3N | 80 | 72 | 10 |

This result turned our attention toward the use of other reaction conditions. Hence, the influence of base on the catalytic performance of this system was investigated by employing a series of bases in DMF. Comparisons between the various bases utilized showed that organic bases are more effective than inorganic bases like K2CO3 and K3PO4. Due to the optimization parameters, DMF and Bu3N were chosen as the most suitable solvent and base. Under these conditions, the carbonylated product was obtained in higher yield and the ether side-product formation was reduced to 6% (Table 1, entry 6). Therefore, we decided that the application of a base was necessary for the reaction to complete. The temperature effect on the reaction was also studied. No serious decrease in the yield of the desired product was observed when the reaction temperature was decreased from 100 °C to 80 °C (Table 1, entry 7). As regards even lower temperatures, the carbonylated product was achieved in lower yields; therefore, 80 °C was considered as the optimum reaction temperature for further studies. The effect of catalyst loading was also examined. It was observed that the yield of the product was decreased with lower loading values of the catalyst (Table 1, entry 9). Similarly, by reducing the amount of alcohol, the yield of the product decreased (Table 1, entry 10).
In order to investigate the effect of substituent groups, a variety of aryl iodides were tested in the reaction with different alcohols, and the desired aryl esters were obtained in good to excellent yields. Scheme 3 clearly reveals that the prepared catalyst is effective for the alkoxycarbonylation reaction of the tested variety of aryl iodides.
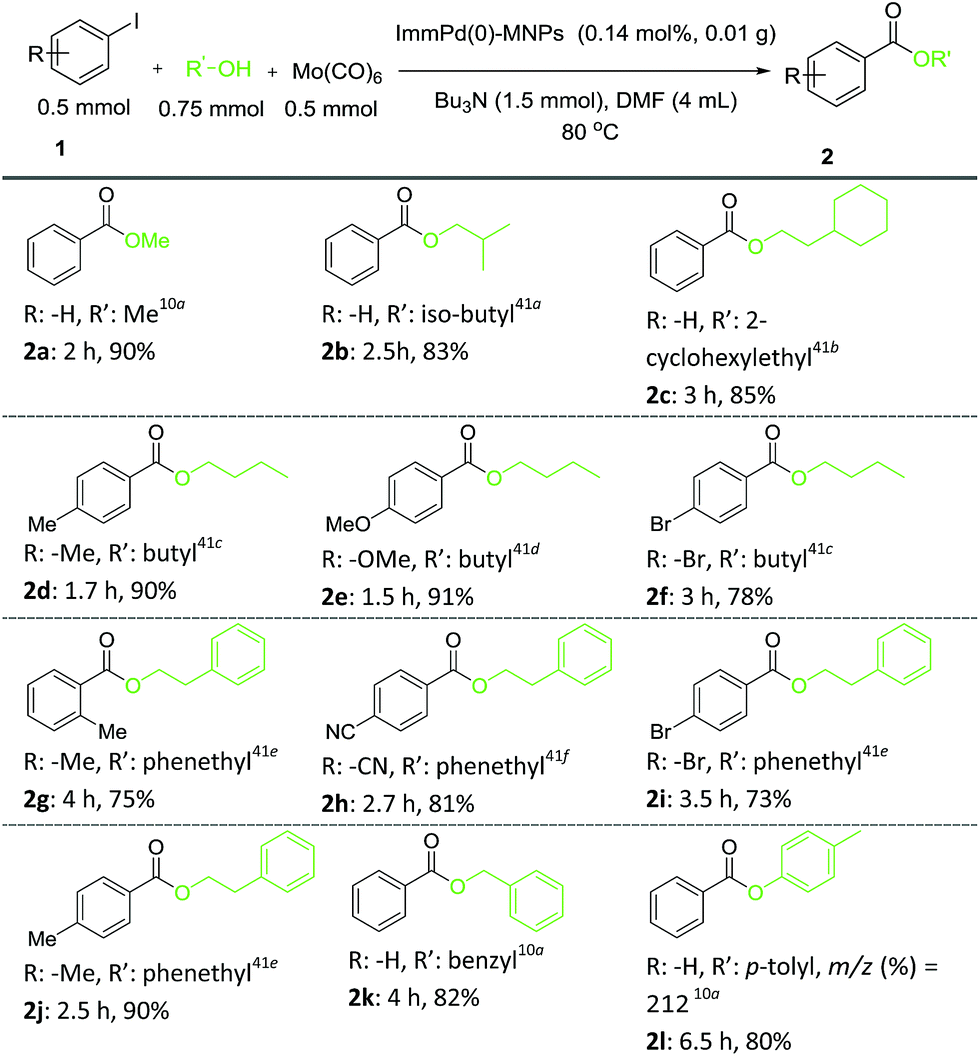 | ||
| Scheme 3 ImmPd(0)-MNPs-catalyzed alkoxycarbonylation of aryl iodides in DMF at 80 °C. | ||
With regard to the results shown in Scheme 3, the carbonylation reaction of aryl iodides bearing either electron-donating or electron-withdrawing substituents afforded the corresponding carbonylative products in good to excellent yields. Strong electron-donating groups, such as the methyl and methoxy group on the phenyl ring of the aryl iodides, resulted in slightly higher yields than for the aryl iodides with electron-withdrawing groups, such as CN and Br (Scheme 3). The differences in the yield of the product of methyl, methoxy, and CN groups can be attributed to the different efficiencies of separation.
The relative position of the substituents had a slight influence on the coupling efficiency. As a representative of sterically hindered substrates, 2-iodotoluene resulted in a lower yield than its p-analog (3fe′, Scheme 3). Furthermore, this catalytic system could be applied to the reaction of phenols with iodobenzene to give the corresponding esters in high yields (3ag′, Scheme 3). The analogous less expensive aryl bromides or chlorides are more attractive substrates; however, the reaction often needs harsher reaction conditions. We examined the use of aryl bromides to synthesize the aryl esters under the optimized reaction conditions, but only a trace amount of yield was obtained. Aryl chloride did not react under the optimized reaction conditions.
Aminocarbonylation of aryl iodides catalyzed by ImmPd(0)-MNPs
The applicability of ImmPd(0)-MNPs toward aminocarbonylation reactions were evaluated using the conditions previously employed for alkoxycarbonylation, that is, ImmPd(0)-MNPs (0.14 mol%), Bu3N (1.5 mmol), 0.5 mmol of amine, 4 mL of DMF at 80 °C in the presence of Mo(CO)6. First, iodobenzene was subjected to the aminocarbonylation reaction with morpholine under these conditions. The results obtained from the aminocarbonylation reaction indicated that these conditions were not effective and only provided the corresponding amide (6aa′) in a 60% yield (Table 2, entry 1). Therefore, we changed the reaction conditions. Further experiments demonstrated that toluene was the best solvent (Table 2, entry 2).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Base | T/°C | 4a (%) |
| a Reaction conditions: iodobenzene (0.5 mmol), morpholine (0.5 mmol), base (1.5 mmol), catalyst (0.01 g, 0.14 mol%), Mo(CO)6 (0.5 mmol), solvent (4 mL), time (1 h). | ||||
| 1 | DMF | Bu3N | 80 | 60 |
| 2 | Toluene | Bu3N | 80 | 95 |
| 3 | Toluene | K2CO3 | 80 | 51 |
| 4 | Toluene | Bu3N | 60 | 40 |
The best optimized reaction condition was then applied to various substituted aryl iodides using 0.14 mol% of catalyst and Mo(CO)6 as a solid source of CO at 80 °C in toluene (Scheme 4). Moreover, various amines were well tolerated to give the desired amides in good to excellent yields (Scheme 4). Both primary and secondary amines provided the desired amide in high yields. It is significant that the amidation of aryl iodides with benzylamine employing ImmPd(0)-MNPs was performed successfully with no additional N-arylation side-products under the optimal conditions.
 | ||
| Scheme 4 ImmPd(0)-MNPs-catalyzed aminocarbonylation of aryl iodides in toluene at 80 °C. | ||
Moreover, in the case of aromatic amines such as aniline, the carbonylated product was formed in a good yield (3af′, 3ag′, Scheme 4).
Catalyst reusability
The level of reusability is an important point concerning the use of heterogeneous catalysts. Easy catalyst separation and recycling in sequential set operations can significantly increase the efficiency of the reaction. We examined the reusability of the present heterogeneous palladium catalyst in the carbonylation of iodobenzene with methanol under the optimized reaction conditions. After completion of the reaction, the catalyst was separated by an external magnet, washed with water and acetone (3 × 5 mL), and then dried at room temperature to be used in the next run. The recovered catalyst could be reused eight times with no significant decrease observed in the activity of the ImmPd(0)-MNPs catalyst (Fig. 8). In order to investigate that the observed catalytic activity is from the supported palladium nanoparticles on the Fe3O4–CS–methionine and not from leached Pd in the solution, a hot-filtration test was performed in the carbonylation of iodobenzene. Here, after continuing the reaction for 1 h, the catalyst was removed by an external magnet and the determined conversion was 60%; the resulting filtrate was then heated for a further 6 h, and it was found that after separation of the catalyst no conversion occurred in the filtrate part. | ||
| Fig. 8 Catalyst reusability test of the ImmPd(0)-MNPs catalyst. | ||
To determine this topic, TEM images and ICP analyses were performed for the recycled catalyst to study the possible changes in Pd particle morphology/size and Pd content on the catalyst surface in comparison with fresh catalyst. As shown in Fig. 1, the TEM image of the catalyst illustrates that the morphology and size of the catalyst does not change considerably after recycling eight times. Additionally, the ICP analysis data of the recovered catalyst after recycling eight times indicates that during the reaction process only a small amount of Pd nanoparticles (0.2 ppm) is lost. With regard to these results, we concluded that the high catalytic activity is related to the ImmPd(0)-MNPs catalyst and not from the leached palladium.
Experimental
Chemical, instrumentation, and analysis
All the chemical reagents were purchased from Merck and Aldrich Chemical Companies and were used without further purification. Chitosan was purchased from Acros Organics and its molecular weight was 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–300000. 1H-NMR and 13C-NMR spectra were recorded on a Bruker (250 MHZ) Avance DRX in pure CDCl3 solvent with tetramethylsilane (TMS) as the internal standard. FT-IR spectra were obtained as KBr pellets on a JASCO 680 Plus spectrophotometer for characterization of the ImmPd-MNPs catalyst. Magnetic measurements were done using a vibrating sample magnetometer (VSM) in 0.0001–50 emu. Transmission electron microscopy (TEM) using a TEM device (Zeiss-EM10C-80 kV) and field emission scanning electron microscopy [MIRA3 TESCAN] were acquired for characterization of the ImmPd-MNPs catalyst. The thermogravimetry analysis (TGA) of the catalyst was studied using a lab-made TGA instrument. The X-ray diffraction (XRD, D8, Advance, Bruker, axs) was engaged for characterization of the ImmPd-MNPs catalyst. The reaction monitoring was accomplished by TLC or gas chromatography (GC) (BEIFIN 3420 gas chromatograph equipped with a Varian CP SIL 5CB column: 30 m, 0.32 mm, 0.25 mm). Column chromatography was carried out on columns of silica gel 60 (70–230 mesh).
000–300000. 1H-NMR and 13C-NMR spectra were recorded on a Bruker (250 MHZ) Avance DRX in pure CDCl3 solvent with tetramethylsilane (TMS) as the internal standard. FT-IR spectra were obtained as KBr pellets on a JASCO 680 Plus spectrophotometer for characterization of the ImmPd-MNPs catalyst. Magnetic measurements were done using a vibrating sample magnetometer (VSM) in 0.0001–50 emu. Transmission electron microscopy (TEM) using a TEM device (Zeiss-EM10C-80 kV) and field emission scanning electron microscopy [MIRA3 TESCAN] were acquired for characterization of the ImmPd-MNPs catalyst. The thermogravimetry analysis (TGA) of the catalyst was studied using a lab-made TGA instrument. The X-ray diffraction (XRD, D8, Advance, Bruker, axs) was engaged for characterization of the ImmPd-MNPs catalyst. The reaction monitoring was accomplished by TLC or gas chromatography (GC) (BEIFIN 3420 gas chromatograph equipped with a Varian CP SIL 5CB column: 30 m, 0.32 mm, 0.25 mm). Column chromatography was carried out on columns of silica gel 60 (70–230 mesh).
Catalyst preparation
General procedure for alkoxycarbonylation in the presence of the ImmPd-MNPs catalyst
In a typical experiment, to a flask containing a mixture of aryl iodide (0.5 mmol), alcohol (0.75 mmol), Bu3N (1.5 mmol, 0.35 mL), and Mo(CO)6 (0.5 mmol, 0.123 g) in 4 mL DMF, ImmPd(0)-MNPs catalyst (0.01 g, 0.14 mol%) was added and stirred at 80 °C for the time specified in Scheme 3. The reaction was monitored by TLC or GC. After the consumption of aryl iodide, the mixture was cooled down to room temperature and the catalyst was removed by an external magnet. Then, H2O (10.0 mL) was added to the remaining solution and the resulting mixture was extracted with EtOAc (3 × 5 mL) and dried over Na2SO4. The products were purified by column chromatography (hexane/ethyl acetate) to achieve the desired purity.Spectral data for the selected esters
General procedure for aminocarbonylation in the presence of the ImmPd-MNPs catalyst
A mixture of aryl iodide (0.5 mmol), amine (0.5 mmol), Bu3N (1.5 mmol, 0.35 mL), Mo(CO)6 (0.5 mmol, 0.123 g), and Pd-catalyst (0.14 mol%, 0.01 g) was stirred in toluene (4.0 mL) at 80 °C in a round-bottom flask. TLC analysis was used to control the completion of the reaction. After cooling, the catalyst was removed by an external magnet and the desired compound was purified by chromatography on silica gel using n-hexane/EtOAc (20/5) as the eluent to afford the corresponding amide in a high yield.Spectral data for the selected amides
Conclusions
In conclusion, we developed and characterized a magnetic chitosan-supported Pd(0) complex and tested its successful application in the alkoxycarbonylation and aminocarbonylation of aryl iodides with alcohols and amines in the presence of Mo(CO)6. Both electron-rich and electron-deficient aryl iodides could be applied for the transformation. The ImmPd(0)-MNPs catalyst was very stable and could be reused easily following extraction by an external magnet without activity loss. Moreover, the present catalyst could be synthesized readily from inexpensive and commercially available starting materials. We believe that this catalyst has great potential for additional appropriate applications in other palladium transformations in the future.Acknowledgements
We gratefully acknowledge the funding support received for this project from the Isfahan University of Technology (IUT), IR Iran (A.R.H.) and Grant GM 33138 (A.E.R.) from the National Institutes of Health, USA. Further financial support from the Center of Excellence in Sensor and Green Chemistry Research (IUT) is gratefully acknowledged.Notes and references
- A. Brennfuhrer, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2009, 48, 4114 CrossRef PubMed.
- (a) X. F. Wu, H. Neumann and M. Beller, Chem. Soc. Rev., 2011, 40, 4986 RSC; (b) M. Seayad, J. Seayad, P. L. Mills and R. V. Chaudhari, Ind. Eng. Chem. Res., 2003, 42, 2496 CrossRef; (c) S. P. Gupte, V. P. Krishnamurthy and R. V. Chaudhari, Chem. Eng. Sci., 1996, 51, 2069 CrossRef CAS.
- H. Neumann, A. Brennfuhrer and M. Beller, Chem.–Eur. J., 2008, 14, 3645 CrossRef CAS PubMed.
- Z. Zhang, Y. Liu, M. Gong, X. Zhao, Y. Zhang and J. Wang, Angew. Chem., Int. Ed., 2010, 49, 1139 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann, A. Spannenberg, T. Schulz, H. Jiao and M. J. Beller, J. Am. Chem. Soc., 2010, 132, 14596 CrossRef CAS PubMed.
- Q. Liu, G. Li, J. He, J. Liu, P. Li and A. Lei, Angew. Chem., Int. Ed., 2010, 49, 3371 CrossRef CAS PubMed.
- X. F. Wu, H. Neumann and M. Beller, Angew. Chem., Int. Ed., 2010, 49, 5284 CrossRef CAS PubMed.
- N. Iranpoor, H. Firouzabadi, E. Etemadi-Davan, A. Nematollahi and H. R. Firouzi, New J. Chem., 2015, 39, 6445 RSC.
- T. Sugihara, C. Coperet, Z. Owczarcy, L. S. Haring and E. J. Negishi, J. Am. Chem. Soc., 1994, 116, 7923 CrossRef CAS.
- (a) M. V. Khedkar, T. Sasaki and B. M. Bhanage, ACS Catal., 2013, 3, 287 CrossRef CAS; (b) V. R. Pattabiraman and J. W. Bode, Nature, 2011, 480, 471 CrossRef CAS PubMed; (c) T. Cupido, J. Tulla-Puche, J. Spengler and F. Albericio, Curr. Opin. Drug Discovery Dev., 2007, 10, 768 CAS; (d) C. Locatelli, F. B. Filippin-Monteiro and T. B. Creczynski-Pasa, Eur. J. Med. Chem., 2013, 60, 233 CrossRef CAS PubMed; (e) A. C. Fonseca, M. H. Gil and P. N. Simoes, Prog. Polym. Sci., 2014, 39, 1291 CrossRef CAS.
- K. Ishihara, Tetrahedron, 2009, 65, 1085 CrossRef CAS.
- M. Beller, B. Cornils, C. D. Frohning and C. W. J. Kohlpaintner, J. Mol. Catal. A: Chem., 1995, 104, 17 CrossRef CAS.
- J. Wannberg and M. J. Larhed, J. Org. Chem., 2003, 68, 5750 CrossRef CAS PubMed.
- H. U. Blaser, M. Diggelmann, H. Meier, F. Naud, E. Scheppach, A. Schnyder and M. J. Studer, J. Org. Chem., 2003, 68, 3725 CrossRef CAS PubMed.
- C. M. Kormos and N. E. Leadbeater, Org. Biomol. Chem., 2007, 5, 65 CAS.
- J. Liu, B. Liang, D. Shu, Y. Hub, Z. Yang and A. Lei, Tetrahedron, 2008, 64, 9581 CrossRef CAS.
- E. Mizushima, T. Hayashi and M. Tanka, Green Chem., 2001, 3, 76 RSC.
- M. A. Mercadante and N. E. Leadbeater, Org. Biomol. Chem., 2011, 9, 6575 CAS.
- C. B. Kelly, C. Lee, M. A. Mercadante and N. E. Leadbeater, Org. Process Res. Dev., 2011, 15, 717 CrossRef CAS.
- K. Kumar, A. Zapf, D. Michalik, A. Tillack, T. Heinrich, H. Bottcher, M. Arlt and M. Beller, Org. Lett., 2004, 6, 7 CrossRef CAS PubMed.
- A. Schoenberg, I. Bartoletti and R. F. Heck, J. Org. Chem., 1974, 39, 3318 CrossRef CAS.
- (a) W. Fang, Q. Deng, M. Xu and T. Tu, Org. Lett., 2013, 15, 3678 CrossRef CAS PubMed; (b) T. T. Dang, Y. Zhu, J. S. Y. Ngiam, S. C. Ghosh, A. Chen and A. M. Seayad, ACS Catal., 2013, 3, 1406 CrossRef CAS; (c) Z. S. Qureshi, S. A. Revankar, M. V. Khedkar and B. M. Bhanage, Catal. Today, 2012, 198, 148 CrossRef CAS; (d) A. S. Prasad and B. Satyanarayana, J. Mol. Catal. A: Chem., 2013, 370, 205 CrossRef; (e) R. H. Munday, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2008, 130, 2754 CrossRef CAS PubMed; (f) H. Zhang, R. Shi, A. Ding, L. Lu, B. Chen and A. Lei, Angew. Chem., Int. Ed., 2012, 51, 12542 CrossRef CAS PubMed; (g) Q. Lin, H. Fu, M. Yuan, H. Chen and X. Li, Acta Phys.-Chim. Sin., 2006, 22, 1272 CrossRef CAS; (h) Y. Yuan, Z. Wang, H. An, W. Xue and Y. Wang, Chin. J. Catal., 2015, 36, 1142 CrossRef CAS.
- (a) K. Yamazak and Y. Kondo, J. Comb. Chem., 2004, 6, 121 CrossRef PubMed; (b) Y. Wan, M. Alterman, M. Larhed and A. Hallberg, J. Org. Chem., 2002, 67, 6232 CrossRef CAS PubMed; (c) B. Roberts, D. Liptrot, L. Alcaraz, T. Luker and M. J. Stocks, Org. Lett., 2010, 12, 4280 CrossRef CAS PubMed; (d) J. Ju, M. Leong, J. Moon, H. M. Jung and S. Lee, Org. Lett., 2007, 9, 4615 CrossRef CAS PubMed; (e) Y. Zhang, H. Sun, W. Zhang, Z. Gao, P. Yang and J. Gu, Appl. Catal., A, 2015, 496, 9 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi, S. Motevalli and M. Talebi, Tetrahedron, 2013, 69, 418 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi and S. Motevalli, J. Mol. Catal. A: Chem., 2012, 355, 69 CrossRef CAS.
- N. Iranpoor, H. Firouzabadi, Z. Tavangar-Rizi and S. Erfan, RSC Adv., 2014, 4, 43178 RSC.
- (a) W. Magerlein, A. F. Indolese and M. J. Beller, Organomet. Chem., 2002, 641, 30 CrossRef CAS; (b) A. Schoenberg and R. F. J. Heck, J. Org. Chem., 1974, 39, 3327 CrossRef CAS; (c) P. W. Miller, N. J. Long, A. J. Mello, R. Vilar, J. Passchier and A. Gee, Chem. Commun., 2006, 5, 546 RSC; (d) J. R. Martinelli, T. P. Clark, D. A. Watson, R. H. Munday and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 8460 CrossRef CAS PubMed; (e) J. R. Martinelli, D. A. Watson, D. M. M. Freckmann, T. E. Barder and S. L. J. Buchwald, J. Org. Chem., 2008, 73, 7102 CrossRef CAS PubMed.
- (a) J. Liu, J. Chen and C. Xia, J. Catal., 2008, 253, 56 CrossRef; (b) P. J. Tambade, Y. P. Patil, M. J. Bhanushali and B. M. Bhanage, Tetrahedron Lett., 2008, 49, 2224 CrossRef; (c) M. V. Khedkar, P. J. Tambade, Z. S. Qureshi and B. M. Bhanage, Eur. J. Org. Chem., 2010, 6986 Search PubMed; (d) M. V. Khedkar, S. R. Khan, D. N. Sawant, D. B. Bagal and B. M. Bhanage, Adv. Synth. Catal., 2011, 353, 3415 CrossRef CAS; (e) S. T. Gadge, M. V. Khedkar, S. R. Lanke and B. M. Bhanage, Adv. Synth. Catal., 2012, 1, 354 Search PubMed; (f) S. P. Chavan and B. M. Bhanage, Tetrahedron Lett., 2014, 55, 1199 CrossRef CAS; (g) S. T. Gadge and B. M. Bhanage, J. Org. Chem., 2013, 78, 6797 CrossRef PubMed; (h) R. S. Mane and B. M. Bhanage, RSC Adv., 2015, 5, 76122 RSC.
- (a) M. Z. Cai, C. S. Song and X. Huang, Synth. Commun., 1997, 27, 366 Search PubMed; (b) M. Cai, Y. Huang, R. Hu and C. Song, J. Mol. Catal. A: Chem., 2004, 212, 154 CrossRef; (c) M. Papp and R. Skoda-Foldes, J. Mol. Catal. A: Chem., 2013, 378, 199 CrossRef; (d) M. V. Khedkar, A. R. Shinde, T. Sasaki and B. M. Bhanage, J. Mol. Catal. A: Chem., 2014, 385, 97 CrossRef; (e) S. M. Lu and H. Alper, J. Am. Chem. Soc., 2005, 9, 14777 Search PubMed.
- M. Cai, J. Peng, W. Hao and G. Ding, Green Chem., 2011, 13, 196 RSC.
- (a) A. Mansour and M. Portnoy, J. Mol. Catal. A: Chem., 2006, 250, 43 CrossRef; (b) S. M. Islam, K. Ghosh, A. S. Roy and R. A. Molla, RSC Adv., 2014, 4, 38986 RSC; (c) S. M. Islam, R. A. Molla, A. S. Roy and K. Ghosh, RSC Adv., 2014, 4, 26181 RSC; (d) Y. Lei, L. Wu, X. Zhang, M. Hui, Y. Gu and G. Li, J. Mol. Catal. A: Chem., 2015, 398, 169 CrossRef.
- (a) M. Genelot, V. Dufaudm and L. Djakovitch, Adv. Synth. Catal., 2013, 355, 2616 CrossRef; (b) M. Genelot, N. Villandier, A. Bendjeriou, P. Jaithong, L. Djakovitch and V. Dufaud, Catal. Sci. Technol., 2012, 2, 1886 RSC.
- T. T. Dang, Y. Zhu, S. C. Ghosh, A. Chen, C. L. L. Chai and A. M. Seayad, Chem. Commun., 2012, 48, 1807 Search PubMed.
- R. S. Mane, T. Sasaki and B. M. Bhanage, RSC Adv., 2015, 5, 94776 RSC.
- R. Rahimi, A. Maleki, A. Morsali and M. J. Rahimi, Solid State Sci., 2014, 28, 9 CrossRef CAS.
- A. A. Galhoum, M. G. Mafhouz, S. T. Abdel-Rehem, N. A. Gomaa, A. A. Atia, T. Vincent and E. Guibal, Nanomaterials, 2015, 5, 154 CrossRef CAS.
- J. Zhou, Z. Dong, H. Yang, Z. Shi, X. Zhou and R. Li, Appl. Surf. Sci., 2013, 279, 360 CrossRef CAS.
- (a) X. Zhang, C. Jiao, J. Wang, Q. Liu, R. Li, P. Yang and M. Zhang, Chem. Eng. J., 2012, 198, 412 CrossRef; (b) M. Namdeo and S. K. Bajpai, Colloids Surf., A, 2008, 320, 161 CrossRef CAS.
- A. Naghipour and A. Fakhri, Catal. Commun., 2016, 73, 39 CrossRef CAS.
- (a) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 1036 CrossRef CAS; (b) A. M. Sajith and A. Muralidharan, Tetrahedron Lett., 2012, 53, 5206 CrossRef CAS; (c) J. F. Hartwig, Inorg. Chem., 2007, 46, 1936 CrossRef CAS PubMed; (d) V. F. Slagt, A. H. M. De Vries, J. G. De Vries and R. M. Kellogg, Org. Process Res. Dev., 2010, 14, 30 CrossRef CAS; (e) P. Mondal, S. Banerjee, A. S. Roy, T. K. Mandal and S. M. Islam, J. Mater. Chem., 2012, 22, 20434 RSC; (f) S. M. Islam, P. Mondal, A. S. Roy, S. Mondal and D. Hossain, Tetrahedron Lett., 2010, 51, 2067 CrossRef CAS.
- (a) R. A. Flath, T. R. Mon, G. Lorenz, C. James Whitten and J. W. Mackley, J. Agric. Food Chem., 1983, 31, 1167 CrossRef CAS; (b) J. Salvadori, E. Balducci, S. Zaza, E. Petricci and M. Taddei, J. Org. Chem., 2010, 75, 1841 CrossRef CAS PubMed; (c) S. Samanta, V. Pappula, M. Dinda and S. Adimurthy, Org. Biomol. Chem., 2014, 12, 9453 RSC; (d) J. Georgsson, A. Hallberg and M. Larhed, J. Comb. Chem., 2003, 5, 350 CrossRef CAS PubMed; (e) C. Zhang, P. Feng and N. Jiao, J. Am. Chem. Soc., 2013, 135, 15257 CrossRef CAS PubMed; (f) X. Huang, X. Li, M. Zou, S. Song, C. Tang, Y. Yuan and N. Jiao, J. Am. Chem. Soc., 2014, 136, 14858 CrossRef CAS PubMed.
- (a) K. Kondo, E. Sekimoto, J. Nakao and Y. Murakami, Tetrahedron, 2000, 56, 5843 CrossRef CAS; (b) D. A. Ockey, J. L. Dotson, M. E. Struble, J. T. Stults, J. H. Bourell, K. R. Clark and T. R. Gadek, Bioorg. Med. Chem., 2004, 12, 37 CrossRef CAS PubMed; (c) G. L. Thomas, C. Böhner, M. Ladlow and D. R. Spring, Tetrahedron, 2005, 61, 12153 CrossRef CAS; (d) I. Mohammadpoor-Baltork, H. R. Memarian and K. Bahrami, Monatsh. Chem., 2004, 135, 411 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18679c |
| This journal is © The Royal Society of Chemistry 2016 |