
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRh-catalyzed direct synthesis of 2,2′-dihydroxybenzophenones and xanthones†
Maddali L. N.
Rao
* and
Boddu S.
Ramakrishna
Department of Chemistry, Indian Institute of Technology Kanpur, Kanpur-208016, India. E-mail: maddali@iitk.ac.in; Fax: +91 512 259 7532; Tel: +91 512 259 7532
First published on 9th August 2016
Abstract
An efficient rhodium-catalyzed direct synthesis of 2,2′-dihydroxybenzophenones and xanthones was developed from functionalized salicylaldehydes. This approach provides an easy access to various functionalized 2,2′-dihydroxybenzophenone and xanthone core skeletons. This study also revealed the crucial role of the hydroxy group in the reductive homo-coupling process to generate 2,2′-dihydroxybenzophenones. Overall the outcome of the reaction course was also found to be influenced by the electronics of the substituent groups in salicylaldehydes.
Introduction
Dihydroxybenzophenone is a medicinally important structural motif with various biological activities.1,2 Some of the derivatives have been used as sunscreen additives3 and as intermediates in the synthesis of chiral ligands.4 Furthermore, symmetrically substituted xanthone derivatives5 have been used as precursors in the synthesis of fluorophores5a,b and in fluorescent dyes.5c Additionally, xanthone such as persulfated 3,6-(O-β-glucopyranosyl)xanthone (E) was known as dual inhibitor of FXa (anticoagulant agent) and with antiplatelet effect.6 Some of the relevant 2,2′-dihydroxybenzophenone and xanthone skeletons are given in Fig. 1.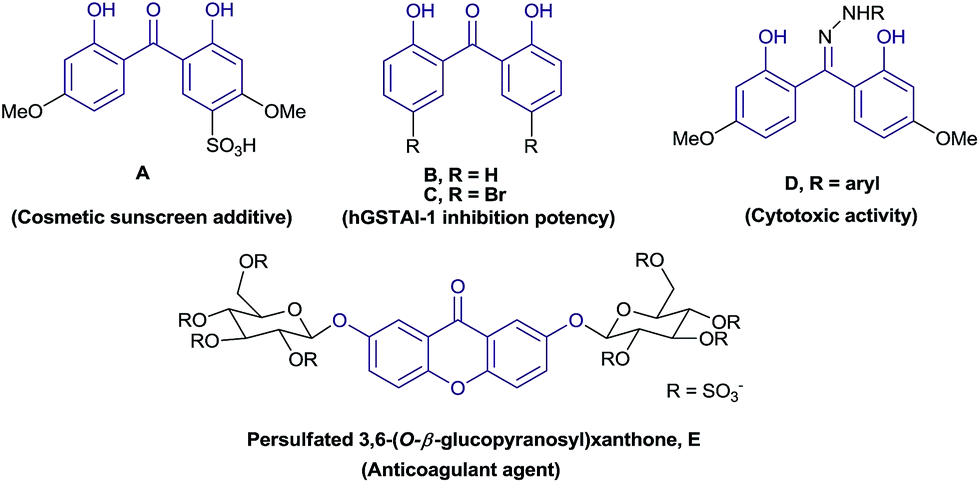 | ||
| Fig. 1 Biologically active molecules. | ||
In this context, methods involving the synthesis of 2,2′-dihydroxybenzophenones are noteworthy and rely on the Fries rearrangement of O-acylphenols,7 Lewis acid promoted arylation of phenols8 and hydrolysis of xanthone derivatives.1a In addition, some of the metal-catalyzed approaches known for the synthesis of 2,2′-dihydroxybenzophenones and xanthones are given in Scheme 1. In literature, the direct hydroxylation of ortho-selective C–H oxygenations involving Pd, Cu, Ru or Rh catalysts were reported for the synthesis of 2,2′-dihydroxybenzophenones.9–11
 | ||
| Scheme 1 Synthesis of 2,2′-dihydroxybenzophenone and xanthone. | ||
Li and co-workers reported one-step synthesis of xanthone derivatives from aryloxybenzaldehydes involving cross-dehydrogenative coupling process.12 Furthermore, Peng and co-workers reported the synthesis of xanthones from 2-hydroxybenzaldehyde and 1,2-dibromoarene under Pd-catalyzed conditions.13 Further, 2-hydroxybenzophenone derivatives with suitable functional groups such as bromo, methoxy and nitro groups were used for the synthesis of xanthones in the presence of copper catalyst.14 Further, Li group also showed the role of various phosphine ligands in the oxidative decarbonylation of aromatic aldehydes to give biaryls and arylketones.15 However, this reactivity with ortho-substituted aromatic aldehydes was found to be non-selective. In addition, the study of aromatic aldehydes with ortho-hydroxy substitution i.e. salicylaldehydes is conspicuously missing in the literature. However, in some instances ortho-hydroxy group was known to retard decarbonylation of aromatic aldehydes under rhodium-catalyzed conditions16–19 and this could be one reason for the lack of study with salicylaldehydes in decarbonylative couplings. These substrates were effectively used in hydroacylations of activated alkenes16,18 and alkynes.17 Against this background, in our recent study with salicylaldehydes under rhodium-catalyzed conditions, we found the novel reductive decarbonylative couplings of salicylaldehydes to give symmetrically substituted 2,2′-dihydroxybenzophenones and xanthones selectively under different reaction conditions (Scheme 1). It is our pleasure to report these results.
Results and discussion
In our study using salicylaldehyde (1a) under rhodium-catalyzed conditions (Table 1), initially we observed the formation of 2,2′-dihydroxybenzophenone (2.1) in 52% yield (entry 1). This prompted us to standardize the condition for the direct synthesis of 2,2′-dihydroxybenzophenone from salicylaldehyde.|
|
|||||
|---|---|---|---|---|---|
| Entry | Catalyst | Base | Additive (equiv.) | Solvent | Yield 2.1 (%) |
| a Conditions: salicylaldehyde (0.5 mmol, 1 equiv.), catalyst (0.025 mmol, 0.05 equiv.), base (1 mmol, 2 equiv.), additive, 100 °C, solvent (3 mL), 16 h. b Isolated yields. | |||||
| 1 | Rh(CO)2(acac) | Na2CO3 | — | DMF | 52 |
| 2 | Rh(CO)2(acac) | Na2CO3 | — | DMA | 28 |
| 3 | Rh(CO)2(acac) | Na2CO3 | — | NMP | 39 |
| 4 | Rh(CO)2(acac) | K3PO4 | — | DMF | 28 |
| 5 | Rh(CO)2(acac) | K2CO3 | — | DMF | 43 |
| 6 | Rh(CO)2(acac) | Na2CO3 | PPh3 (0.1) | DMF | 26 |
| 7 | Rh(CO)2(acac) | Na2CO3 | Et3N (1) | DMF | 48 |
| 8 | Rh(CO)2(acac) | Na2CO3 | TBAB (1) | DMF | 32 |
| 9 | Rh(CO)2(acac) | Na2CO3 | Cu(OAc)2 (1) | DMF | 67 |
| 10 | Rh(CO)2(acac) | Na2CO3 | Cu(OAc)2 (2) | DMF | 80 |
| 11 | RhCl(PPh3)3 | Na2CO3 | Cu(OAc)2 (2) | DMF | 63 |
| 12 | {RhCl(cod)}2 | Na2CO3 | Cu(OAc)2 (2) | DMF | 56 |
| 13 | RhCl3·3H2O | Na2CO3 | Cu(OAc)2 (2) | DMF | 67 |
| 14 | Rh(CO)2(acac) | — | Cu(OAc)2 (2) | DMF | 63 |
| 15 | Rh(CO)2(acac) | — | — | DMF | 24 |
| 16 | — | Na2CO3 | Cu(OAc)2 (2) | DMF | — |

To achieve this, we carried out a thorough systematic screening. For example, this reaction in N,N-dimethylacetamide (DMA) and N-methyl-2-pyrrolidone (NMP) as solvents furnished lower yields of 2,2′-dihydroxybenzophenone (entries 2 and 3). Further screening was done with change in base employing K3PO4 or K2CO3 and these reactions gave 28% and 43% yields respectively (entries 4 and 5). Additional use of triphenylphosphine did not improve the yield (entry 6). A few more additives were screened (entries 7–9) out of which, Cu(OAc)2·H2O (1 equiv.) was found to be relatively more effective giving 67% yield (entry 9). Encouragingly, this yield was further increased to 80% with 2 equiv. of Cu(OAc)2·H2O (entry 10). At this stage, various Rh-catalysts were also screened to test their efficacy (entries 11–13) and this effort proved to be ineffective.
A control reaction carried out without base provided lowered yield (entry 14). Additional controls without base/additive or catalyst gave either poor or no product formation (entries 15 and 16). From this study, it was found that salicylaldehyde (1 equiv.), Rh(CO)2(acac) (0.05 equiv.), Na2CO3 (2 equiv.), Cu(OAc)2·H2O (2 equiv.) at 100 °C in DMF as an effective protocol for the decarbonylative coupling of salicylaldehyde (1a) to give 2,2′-dihydroxybenzophenone (2.1).
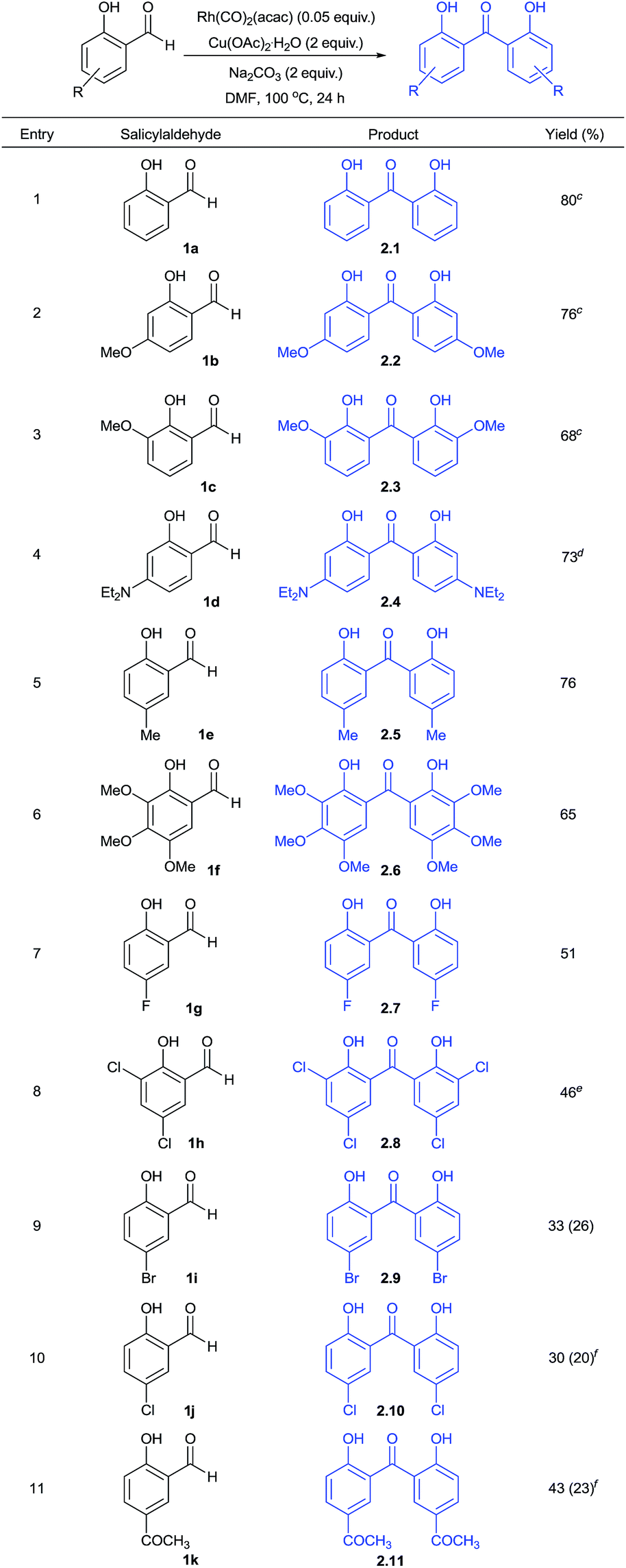
Using this optimized protocol, further scope and reactivity of differently substituted salicylaldehydes was studied (Table 2). This study employing electron-rich salicylaldehydes (1b–1f) was found to be high yielding in comparison to the corresponding reactivity obtained with electron-deficient aryl aldehydes (1g–1k). Thus, salicylaldehyde and other derivatives of it with 4-methoxy, 3-methoxy, 4-N,N-diethylamino, 5-methyl and 3,4,5-trimethoxy groups gave the corresponding functionalized 2,2′-dihydroxybenzophenones in 65–80% yields (2.1–2.6). As given in Fig. 2, the product 2.2 was also confirmed by X-ray analysis.
| a Conditions: salicylaldehyde (0.5 mmol, 1 equiv.), Rh(CO)2(acac) (0.025 mmol, 0.05 equiv.), Na2CO3 (1 mmol, 2 equiv.), Cu(OAc)2·H2O (1 mmol, 2 equiv.), 100 °C, DMF (3 mL), 24 h. b Isolated yields are given. Xanthone product yields are given in parenthesis. c 16 h. d At 120 °C. e 2,4-Dichlorophenol was formed with 10% yield. f Without using Na2CO3. |
|---|
|
 | ||
| Fig. 2 X-ray structure of 2.2. | ||
Moreover, salicylaldehydes substituted with 5-bromo, 5-chloro, 3,5-dichloro, 5-fluoro and 5-acetyl groups gave respective 2,2′-dihydroxybenzophenones in 30–51% yields (2.7–2.11). In some of these cases, we observed the formation of xanthone as a cyclized product in minor amounts (given in the parenthesis, Table 2).
This prompted us to explore whether it is possible to obtain xanthone as a major product directly from salicylaldehydes in a one-pot operation. Hence, we performed this reaction at 100 or 120 °C. These results are summarized in Table 3. The formation of 2,2′-dihydroxybenzophenone and its in situ cyclization to give xanthone was found to be effective under the conditions studied to give moderate yields (3.1–3.10). This effort gave various functionalized xanthones containing formyl, bromo, chloro, fluoro, methyl, acetyl, nitro, hydroxy and trimethoxy groups. One of the positive factors to be highlighted is that salicylaldehydes substituted with electron-withdrawing groups reacted at 100 °C to give the corresponding xanthones in moderate yields. Thus, salicylaldehyde substrates at C-5 substitution with chloro, acetyl, formyl and nitro groups got cyclized to xanthones at 100 °C (3.6–3.9). Other substrates with 5-methyl, 4-hydroxy, 5-fluoro and 3,4,5-trimethoxy groups were cyclized at 120 °C to give xanthone products (3.2–3.4 and 3.10). Further, the obtained reactivity was found to be site-selective as we obtained xanthone 3.8 with formyl group intact using 1l substrate. This indicated the preferential reaction of formyl group ortho to hydroxy during decarbonylative homo-coupling process. The presence of formyl and halo groups in xanthone products endowed them with additional synthetic advantage for further transformations.
| a Conditions: salicylaldehyde (0.5 mmol, 1 equiv.), Rh(CO)2(acac) (0.025 mmol, 0.05 equiv.), Na2CO3 (1 mmol, 2 equiv.), Cu(OAc)2·H2O (1 mmol, 2 equiv.), 120 °C, DMF (3 mL), 24 h. b Isolated yields are given. Yields obtained with 100 °C heating condition are given in parenthesis. c 16 h. d Decarbonylated product phenol formed in minor amount. |
|---|

|
To probe the mechanism, we performed a series of control experiments as given in Scheme 2. Firstly, the reactivity of benzaldehyde and 2-methoxybenzaldehyde was examined under the established conditions and in these cases the corresponding decarbonylative homo-coupled products were not obtained (eqn (1) and (2)). This observation in conjunction with our results with salicylaldehydes indicated that the hydroxy group is crucial for the desired product formation. Next, the reactivity of cross-decarbonylative homo-couplings was studied using two different salicylaldehydes. This led to the formation of a mixture containing both homo- and cross-coupled products (eqn (3)) indicating the involvement of two aldehydic C–H activations at two different stages during the catalytic cycle. Another cross-over experiment performed with salicylaldehyde and p-anisaldehyde gave only homo-coupled product (2.1) from salicylaldehyde in 76% yield (eqn (4)). In this reaction, we have not observed the formation of any cross-coupled product signifying the non-participation of p-anisaldehyde during the reaction course. Thus the presence of –OH group in salicylaldehyde has a definitive role during aldehydic C–H activation.
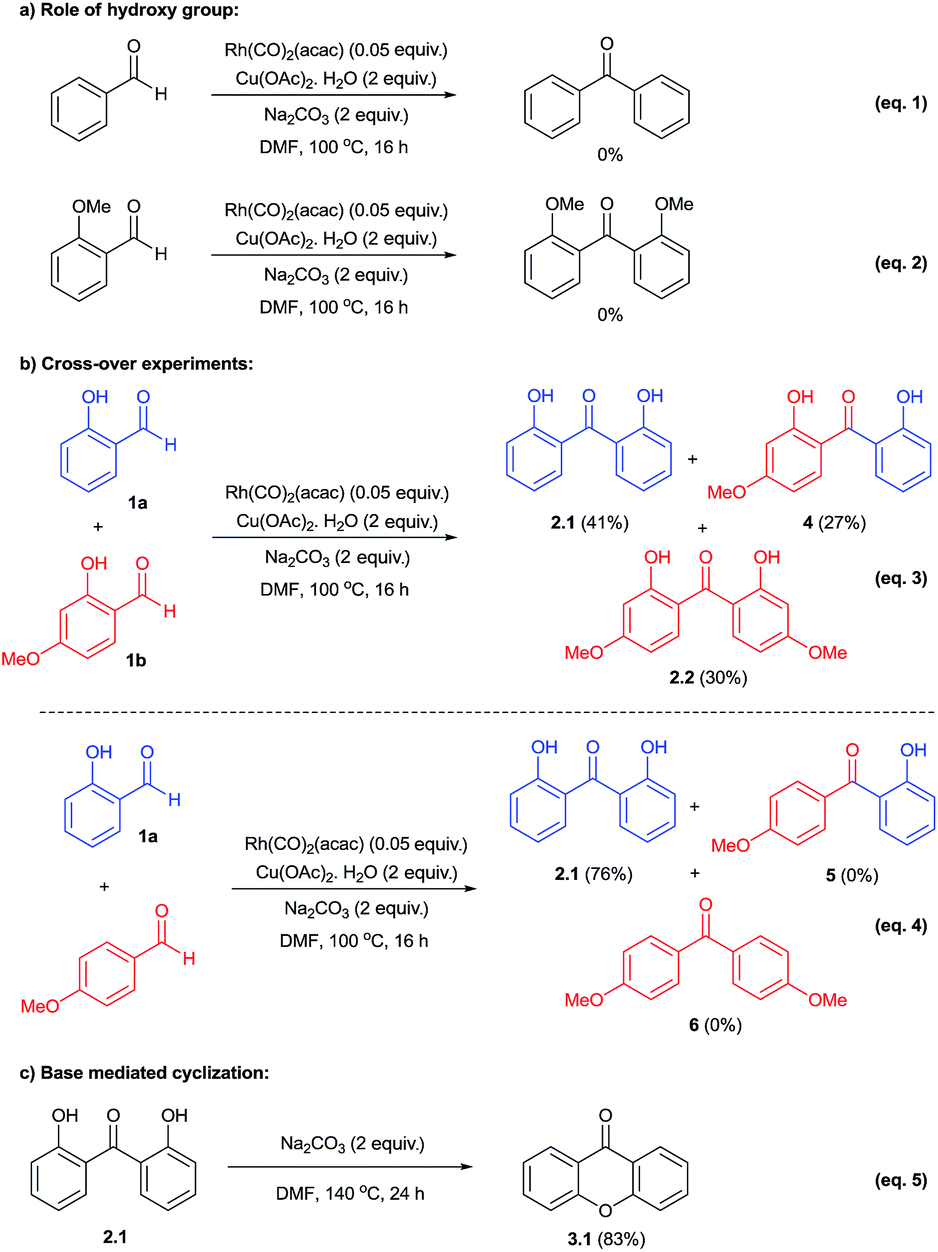 | ||
| Scheme 2 Control experiments. | ||
Further, formation of xanthone was examined with the direct use of 2,2′-dihydroxybenzophenone in the presence of base under heating conditions. This reaction gave xanthone product (3.1) in 83% yield (eqn (5)) and it clearly indicated the important role of base in the cyclization of 2,2′-dihydroxybenzophenone during the reaction course.5c
Based on these investigations, the following mechanistic cycle was proposed (Scheme 3). The initial oxidative addition of salicylaldehyde with Rh(I) was expected to form the intermediate A. This would undergo decarbonylation15 to generate arylrhodium B. This in turn involves in the C–H activation of second molecule of salicylaldehyde followed by copper acetate mediated transformation provides aroylrhodium C species.19 This intermediate would then undergo reductive elimination to give 2,2′-dihydroxybenzophenone (2.1) as decarbonylative homo-coupled product.18 The minor formation of phenol in some cases as decarbonylated product also supports the involvement of arylrhodium B. The proposed second involvement of aldehydic C–H with arylrhodium B also explains the cross-over experiment using two different salicylaldehydes giving a mixture of products (Scheme 2, eqn (3)). The prominent role of –OH indicated by the control reaction carried out by using salicylaldehyde and aryl aldehyde (Scheme 2, eqn (4)) in which case no cross-coupled product (either 5 or 6) was obtained. Further base mediated cyclization of 2,2′-dihydroxybenzophenone directly provided xanthone product under heating conditions (Scheme 2, eqn (5)).5c
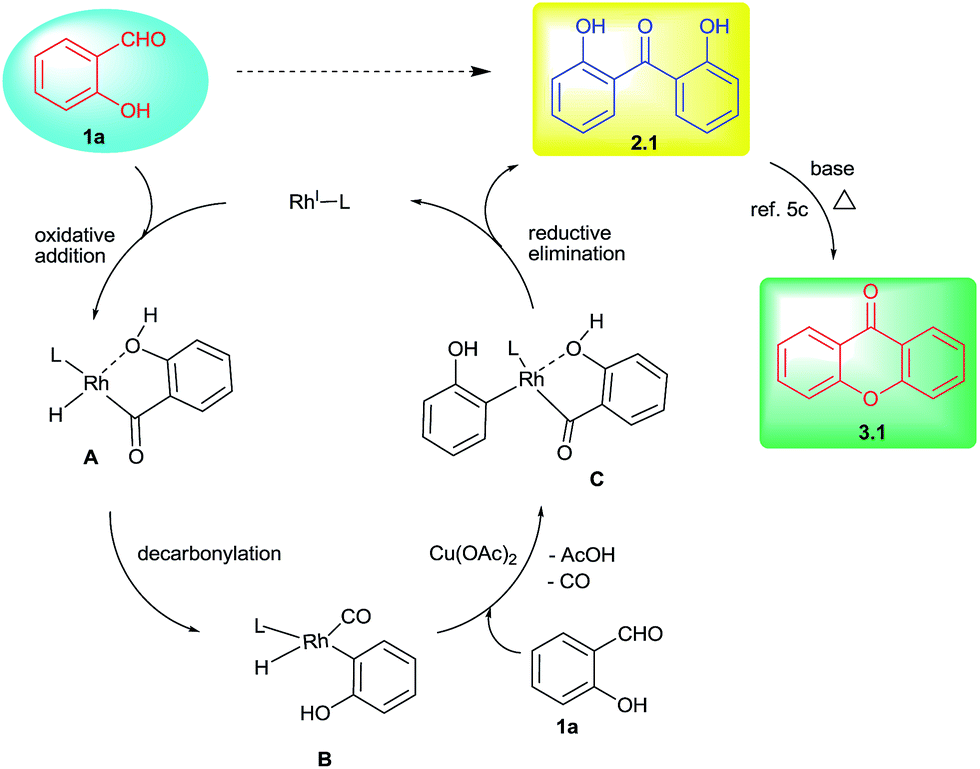 | ||
| Scheme 3 Proposed mechanistic cycle. | ||
Conclusions
In conclusion, we have developed an efficient methodology for the decarbonylative homo-coupling of functionalized salicylaldehydes to give 2,2′-dihydroxybenzophenones. This study was further extended to the preparation of xanthone derivatives directly from functionalized salicylaldehydes in a one-pot operation. Another highlight is that, this study showed the important and crucial role of hydroxy group in the reductive homo-coupling of salicylaldehydes. The overall outcome of the reaction course was also influenced by the electronics of the substituent groups in salicylaldehydes. This study thus unravelled the direct formation of 2,2′-dihydroxybenzophenones and xanthones from functionalized salicylaldehydes under rhodium-catalyzed conditions.Experimental
General
All decarbonylative couplings were performed in dry Schlenk tubes under nitrogen atmosphere conditions. Salicylaldehyde and 3-methoxysalicylaldehyde were purchased from Spectrochem. 5-Bromosalicylaldehyde and 4-N,N-diethylaminosalicylaldehyde were purchased from Sigma-Aldrich. Literature procedures were followed for the preparation of other functionalized salicylaldehydes.20–22 All solvents were dried according to standard procedures. Purification of the products was carried out by silica-gel column chromatography using ethyl acetate/hexane as eluent. 1H and 13C NMR spectra were recorded with JEOL ECS 400 (400 MHz) and JEOL ECX 500 (500 MHz). HRMS was recorded using Electron Ionization (EI) and Electro-Spray Ionization (ESI) techniques with Waters CAB155 GCT Premier analyzer and Waters HAB 213 Q-TOF Premier analyzer. IR spectra were recorded on PerkinElmer FT-IR. X-ray data was recorded on Bruker SMART APEX-II CCD diffractometer. Melting points reported are uncorrected.Representative coupling procedure for Table 2
This reaction was carried out by charging a dry Schlenk tube with salicylaldehyde (61 mg, 0.5 mmol, 1 equiv.), Rh(CO)2(acac) (6.4 mg, 0.025 mmol, 0.05 equiv.), Na2CO3 (106 mg, 1 mmol, 2 equiv.), Cu(OAc)2·H2O (200 mg, 1 mmol, 2 equiv.) and DMF (3 mL). The resultant mixture was stirred at 100 °C in an oil bath for 24 h. After that, contents were cooled to rt, quenched with dil. HCl and extracted with ethyl acetate (30 mL). The organic portion was washed with water (15 mL), brine (15 mL), dried over anhydrous MgSO4 and concentrated. The crude product was subjected to silica-gel column chromatography using ethyl acetate/hexane as eluent. The desired product 2.1 was obtained as yellow liquid (43 mg, 80%).Representative coupling procedure for Table 3
The above procedure given for Table 2 was followed with heating at 120 °C. The desired product 3.1 was obtained as white solid (23 mg, 47%).Acknowledgements
The authors thank the Science and Engineering Research Board (SERB) under Department of Science and Technology (DST), New Delhi for funding this work (SR/S1/OC-20/2012). B. S. R. acknowledges the Council of Scientific and Industrial Research (CSIR), New Delhi for research fellowship.Notes and references
-
(a) F. D. Perperopoulou, P. G. Tsoungas, T. N. Thireou, V. E. Rinotas, E. K. Douni, E. E. Eliopoulos, N. E. Labrou and Y. D. Clonis, Bioorg. Med. Chem., 2014, 22, 3957–3970 CrossRef CAS PubMed
; (b) M. Miyano, J. R. Deason, A. Nakao, M. A. Stealey, C. I. Villamil, D. D. Sohn and R. A. Mueller, J. Med. Chem., 1988, 31, 1052–1061 CrossRef CAS PubMed
-
(a) L. R. Morgan, K. Thangaraj, B. LeBlanc, A. Rodgers, L. T. Wolford, C. L. Hooper, D. Fan and B. S. Jursic, J. Med. Chem., 2003, 46, 4552–4563 CrossRef CAS PubMed
-
(a) S. Tanimoto and A. Toshimitsu, Bull. Inst. Chem. Res., Kyoto Univ., 1991, 69, 560–570 Search PubMed
-
(a) A. Lee and H. Kim, J. Am. Chem. Soc., 2015, 137, 11250–11253 CrossRef CAS PubMed
-
(a) P. Shieh, M. J. Hangauer and C. R. Bertozzi, J. Am. Chem. Soc., 2012, 134, 17428–17431 CrossRef CAS PubMed
- M. Correia-da-Silva, E. Sousa, B. Duarte, F. Marques, F. Carvalho, L. M. Cunha-Ribeiro and M. M. M. Pinto, J. Med. Chem., 2011, 54, 5373–5384 CrossRef CAS PubMed
-
(a) J. A. Miller, J. Org. Chem., 1987, 52, 322–323 CrossRef CAS
- X. Zhang, X. Li, H. Sun, Z. Jiang, L. Tao, Y. Gao, Q. Guo and Q. You, Org. Biomol. Chem., 2012, 10, 3288–3299 CAS
-
(a) G. Shan, X. Yang, L. Ma and Y. Rao, Angew. Chem., Int. Ed., 2012, 51, 13070–13074 CrossRef CAS PubMed
- S.-Z. Sun, M. Shang, H.-L. Wang, H.-X. Lin, H.-X. Dai and J.-Q. Yu, J. Org. Chem., 2015, 80, 8843–8848 CrossRef CAS PubMed
-
(a) K. Kim, H. Choe, Y. Jeong, J. H. Lee and S. Hong, Org. Lett., 2015, 17, 2550–2553 CrossRef CAS PubMed
- P. Wang, H. Rao, R. Hua and C.-J. Li, Org. Lett., 2012, 14, 902–905 CrossRef CAS PubMed
- S. Wang, K. Xie, Z. Tan, X. An, X. Zhou, C.-C. Guo and Z. Peng, Chem. Commun., 2009, 6469–6471 RSC
- J. Hu, E. A. Adogla, Y. Ju, D. Fan and Q. Wang, Chem. Commun., 2012, 48, 11256–11258 RSC
- L. Yang, T. Zeng, Q. Shuai, X. Guo and C.-J. Li, Chem. Commun., 2011, 47, 2161–2163 RSC
-
(a) I. Pernik, J. F. Hooper, A. B. Chaplin, A. S. Weller and M. C. Willis, ACS Catal., 2012, 2, 2779–2786 CrossRef CAS
-
(a) X.-W. Du and L. M. Stanley, Org. Lett., 2015, 17, 3276–3279 CrossRef CAS PubMed
- M. V. Delius, C. M. Le and V. M. Dong, J. Am. Chem. Soc., 2012, 134, 15022–15032 CrossRef PubMed
- Z. Shi, N. Schroder and F. Glorius, Angew. Chem., Int. Ed., 2012, 51, 8092–8096 CrossRef CAS PubMed
-
(a) W. L. Mendelson and S. Hayden, Synth. Commun., 1996, 26, 603–610 CrossRef CAS
-
(a) S. Bhatt and S. K. Nayak, Tetrahedron Lett., 2009, 50, 5823–5826 CrossRef CAS
-
(a) T. Mahajan, L. Kumar, K. Dwivedi and D. D. Agarwal, Ind. Eng. Chem. Res., 2012, 51, 3881–3886 CrossRef CAS
-
(a) F. G. Baddar, L. S. El-Assal and V. B. Baghos, J. Chem. Soc., 1955, 1714–1718 RSC
-
(a) I. Granoth and H. J. Pownall, J. Org. Chem., 1975, 40, 2088–2091 CrossRef CAS
-
(a) S. N. Dhar, J. Chem. Soc., Trans., 1920, 117, 1053–1070 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1480785. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra18647e |
| This journal is © The Royal Society of Chemistry 2016 |