DOI:
10.1039/C6RA18482K
(Paper)
RSC Adv., 2016,
6, 82890-82899
Mirror symmetry breaking in fluorinated bent-core mesogens†
Received
20th July 2016
, Accepted 19th August 2016
First published on 22nd August 2016
Abstract
Spontaneous chirality synchronization in the LC phases of achiral bent-core molecules, the so called dark conglomerate mesophases (DC[*] phases), is a challenging task with significant importance for fundamental scientific research and potential applications. Here we report the synthesis and investigation of two new series of achiral bent-core mesogens derived from 4-bromoresorcinol and 4-chlororesorcinol with 2,3-difluorinated azobenzene-based side arms. The self-assembly of these materials was characterized by DSC, polarizing microscopy, X-ray diffraction investigations (XRD) and electro-optical studies. Depending on the type of halogen substituent at the central resorcinol core and on the terminal alkyl chain length different types of mesophases were observed, where 4-bromoresocinol derived compounds predominately show helical nanocrystallite phases, (HNC phases), representing conglomerates of chiral domains (DC[*]), whereas the related 4-chlororesorcinol based compounds form smectic C phases with a polar domain structure (SmCsPAR). Comparison with related compounds provides information about the influence of core fluorination on the mesophase behaviour and DC[*] phase formation, thus providing a step forward in uncovering the molecular design principles of LC materials capable of mirror symmetry breaking.
1. Introduction
Since the first report by Niori et al.,1 about polar order in liquid crystalline (LC) phases formed by molecules with a non-linear bent shape, the so-called bent-core liquid crystals (BCLCs), extensive research has been done by several research groups on these fascinating materials.2 In recent years BCLCs have received great attention due to their remarkable and unique mesophases, especially the development of macroscopic polar order (ferroelectricity) and spontaneous mirror symmetry breaking, though the molecules themselves are achiral. This phenomenon is of general interest for fundamental soft matter science as well as for potential applications. Mirror symmetry breaking is a basic feature of living matter which was in the recent two decades also observed in some cases of LC phases.2–15 Dark conglomerate phases (DC[*] phases) represent one class of such spontaneously mirror symmetry broken mesophases exhibited by BCLCs.3,16–19 They are optically isotropic and therefore characterized by completely dark appearance between crossed polarizers, whereas under slightly uncrossed polarizers chiral domains of opposite handedness can be observed. Related chiral domains were recently also observed in some nematic phases of dimesogens with odd-numbered spacers (twist bend nematic phases (NTB)),9–11,20 in SmC phases formed by some azobenzene-based BCLCs,21 in bicontinuous cubic phases22 and even in isotropic liquids formed by some polycatenar molecules.6,23,24 Also trimesogen can show NTB phases and soft crystalline DC[*] phases.25 The DC[*] phases of BCLCs are classified into three major types (Fig. 1), including the deformed smectic LC phases with sponge like structure,16,26–36 the helical nano-filament phases (HNF phases or B4 phases) where the molecules are arranged in arrays of helical nano-scale filaments,17,37–44 and the third type of DC[*] phases being the helical nano-crystallites phases (HNC phases) which were recently observed for some azobenzene based BCLCs (see Scheme 1).45–49
 |
| | Fig. 1 The three major types of dark conglomerate (DC[*]) phases.48 | |
 |
| | Scheme 1 Chemical structures of previously reported BCLCs with azobenzene-based wings with and without peripheral fluorine substitution.45–47,49 Abbreviations: Cr = crystalline solid; DC[*] = dark conglomerate phases (HNC phases) composed of chiral domains with opposite handedness; N = nematic phase; SmCPA = antiferroelectric switching smectic phase. | |
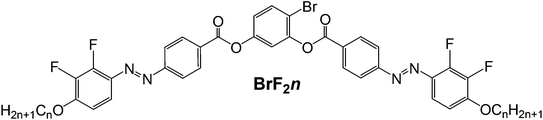
Herein we report two new series of BCLCs derived from 4-bromoresorcinol and 4-chlororesorcinol central cores, respectively,50 with laterally 2,3-difluorinated azobenzene side arms (compounds BrF2n and ClF2n, see Scheme 2). The 4-bromoresorcinol based compounds with medium alkyl chain lengths form helical nanocrystallite phases (HNC phase), representing optically isotropic soft crystalline phases forming conglomerates of chiral domains (DC[*] phases) which for the longest homologue are replaced by a smectic C phase composed of ferroelectric domains with antipolar correlation (SmCsPAR).51 The other series of compounds, which is derived from 4-chlororesorcinol, shows no HNC phases, but exclusively monotropic SmC phases which represent SmCsPAR phases of a slightly different type. These compounds are compared with previously reported compounds without fluorine substitution or with a reduced number of fluorines in the side arms.47,49
 |
| | Scheme 2 Synthetic route to the bent-core mesogens under investigations. | |
2. Experimental
2.1. Synthesis
The synthesis of the target BCLCs BrF2n and ClF2n was carried out as shown in Scheme 2 by acylation reaction of the 4-halogenated resorcinol V with two equivalents of the benzoyl chlorides IVn24 in the presence of triethylamine as base and pyridine as a catalyst. The final crude bent-core compounds BrF2n and ClF2n were purified by column chromatography using dichloromethane followed by recrystallization from ethanol/chloroform (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) mixture to yield the desired materials. Detailed procedures and the analytical data of the newly synthesised compounds BrF2n and ClF2n are reported in the ESI.† All compounds are thermally stable as confirmed by the reproducibility of thermograms in several heating and cooling cycles.
:1) mixture to yield the desired materials. Detailed procedures and the analytical data of the newly synthesised compounds BrF2n and ClF2n are reported in the ESI.† All compounds are thermally stable as confirmed by the reproducibility of thermograms in several heating and cooling cycles.
2.2. Methods
The thermal behaviour of all synthesized compounds was studied by polarizing optical microscopy (POM) and differential scanning calorimetry (DSC). For polarizing microscopy a Mettler FP-82 HT hot stage and control unit in conjunction with a Nikon Optiphot-2 polarizing microscope was used. DSC-thermograms were recorded on a Perkin-Elmer DSC-7 with heating and cooling rates of 10 K min−1. Electro-optical switching characteristics were examined in 6 μm polyimide coated ITO cells (EHC Japan) using the triangular-wave method.52 XRD patterns were recorded with a 2D detector (Vantec-500, Bruker). Ni filtered and pin hole collimated CuKα radiation was used. The exposure time was 15 min and the sample to detector distance was 8.95 and 26.7 cm for small angle and wide angle scattering experiments, respectively. Alignment was attempted by slow cooling (rate: 1 K min−1 to 0.1 K min−1) of a small droplet on a glass plate.
3. Results and discussion
3.1. Dark conglomerate phases of compounds BrF2n with n ≤ 14
Depending on the terminal chain length, compounds BrF2n with a 4-bromoresorcinol central core form different types of mesophases. The shortest derivative BrF28 with n = 8 forms a birefringent crystalline solid with melting point T = 104 °C, whereas compounds BrF210–BrF214 with medium chain length exhibit monotropic highly viscous optically isotropic phases which, once formed, do not crystallize even after storage for one year at room temperature (see Table 1). Under crossed polarizers, these isotropic phases appear completely dark between crossed polarizers and on rotating the analyzer by a small angle (∼10°) out of the crossed position i.e. from the 90° position with respect to the polarizer, uniform dark and bright domains appear, indicating the presence of chiral domains (DC[*] phases, see Fig. 2). That the distinct regions represent chiral domains with opposite handedness was confirmed by rotating the sample itself between crossed polarizers in different directions, where no change in the dark texture was observed.
Table 1 Phase transition temperatures (T/°C), mesophase types, and transition enthalpies [ΔH/kJ mol−1] of compounds BrF2na
|
|
| Compound |
n
|
Heating T/°C [ΔH/kJ mol−1] |
Cooling T/°C [ΔH/kJ mol−1] |
|
The phase transition temperatures (peak temperatures) were taken from the second heating and second cooling scans at 10 K min−1; abbreviations: Cr = crystalline solid; DC[*] = dark conglomerate phases (HNC phases) composed of chiral domains with opposite handedness; SmCsPAR = polarization randomized smectic phase composed of SmCsPF domains and showing two polarization current peaks; Iso = isotropic liquid.
|
|
BrF28
|
8 |
Cr1 96 [6.6] Cr2 104 [27.5] Iso |
Iso 86 [28.5] Cr |
|
BrF210
|
10 |
Cr 101 [37.6] Iso |
Iso 85 [20.4] DC[*] |
|
BrF212
|
12 |
DC[*] 97 [10.1] Cr 103 [10.9] Iso |
Iso 90 [21.0] DC[*] |
|
BrF214
|
14 |
DC[*] 98 [11.9] Cr 103 [7.8] Iso |
Iso 92 [21.6] DC[*] |
|
BrF216
|
16 |
Cr 97 [26.5] Iso |
Iso 96 [6.2] SmCsPAR 85 [20.2] Cr |
 |
| | Fig. 2 Textures of the DC[*] phase of compound BrF212 at T = 60 °C: (b) under crossed polarizers; (a) after rotating the analyzer by 10° from the crossed position with respect to the polarizer in clock-wise direction and (c) in anticlockwise direction, showing dark and bright domains, indicating the presence of areas with opposite chirality sense. | |
The DC[*]-Iso transitions are associated with transition enthalpies values around ΔH ∼ 21 kJ mol−1 in the cooling cycles (see Fig. 3 and Table 1), similar to their related analogues with only one fluorine substituent in each of the outer rings of the bent-core structure (compounds BrFn in Scheme 1).47 On heating the DC[*] phases become instable and crystallize with formation of a birefringent crystalline phase. This crystallization is immediately followed by the melting of this crystalline phase, leading to the typical “double peak” in the heating scans (see Fig. 3). Similar to the DC[*] phases exhibited by related BCLCs with two azobenzene wings (IFn, Brn, BrFn and Men, see Scheme 1),45–47,49 no current peak could be observed in these DC[*] phases in electro-optical experiments. Moreover, no birefringence is induced in any of the DC[*] phases under an applied triangular wave voltage up to 200 Vpp in a 6 μm ITO cell; this is typical for soft crystalline DC[*] phases.
 |
| | Fig. 3 DSC heating and cooling scans of BrF212 with a rate of 10 K min−1. | |
The XRD pattern of the DC[*] phase exhibited by compound BrF210 at 50 °C as a representative example is shown in Fig. 4a and b. A single strong scattering in the small angle region is observed. The d-value of 4.33 nm is between half of the molecular length and the full molecular length (Lmol = 5.2 nm in the most extended conformation with all-trans alkyl chains). This diffraction pattern is in line with a lamellar organization with d = 4.33 nm where the molecules are organized in a single layer structure with the involved molecules tilted by a certain angle (the maximum tilt estimated from d/Lmol = 0.83 is ∼33°) with respect to the layer normal. The relatively large difference between d and Lmol is different from HNF phases having very small difference between the d-value and Lmol.17,38 In the 2D patterns all scatterings form closed rings with uniform intensity distribution as a very typical feature of all DC[*] phases. This is due to the randomized orientation of the nanocrystallites, leading to the optical isotropic properties, giving rise to the dark appearance between crossed polarizers (see Fig. 4a). Fig. 4b shows the 2θ scan over the diffraction pattern of BrF210. Beside the strong layer reflections very weak and relatively broad scattering maxima are observed in the medium and wide angle region. This pattern distinguish this DC[*] phase from the fluid sponge-type DC[*] phases, which exhibit only one completely diffuse wide angle scattering besides the layer reflection,26 as well as from the HNF phases (B4 phases) characterized by sharper and more intense wide angle scatterings.17,38 The results obtained for BrF210 prove that the DC[*] phases formed by compounds BrF2n belong to the helical nanocrystallite phases (HNC phases). Only the number, intensities and positions of the medium- and wide-angle scatterings are distinct, indicating that the fine structure of the local crystal lattice should be a bit different from the previously reported HNC phases of the related azobenzene derived apex-halogenated bent-core compounds.45,47,48 For example, Fig. 4c shows the 2θ-scans of the DC[*] phase of the related compound BrF12 with only one lateral fluorine substituent in each azobenzene wing and having the chain length n = 12 (Scheme 1).
 |
| | Fig. 4 (a) 2D XRD pattern of the DC[*] phase of BrF210 at T = 50 °C, the inset shows the small angle region; 2θ-scans over this XRD pattern of the DC[*] phase for (b) compound BrF210 at T = 50 °C and (c) compound BrF12 at T = 90 °C.47 | |
3.2. SmC phase of the long chain compound BrF216
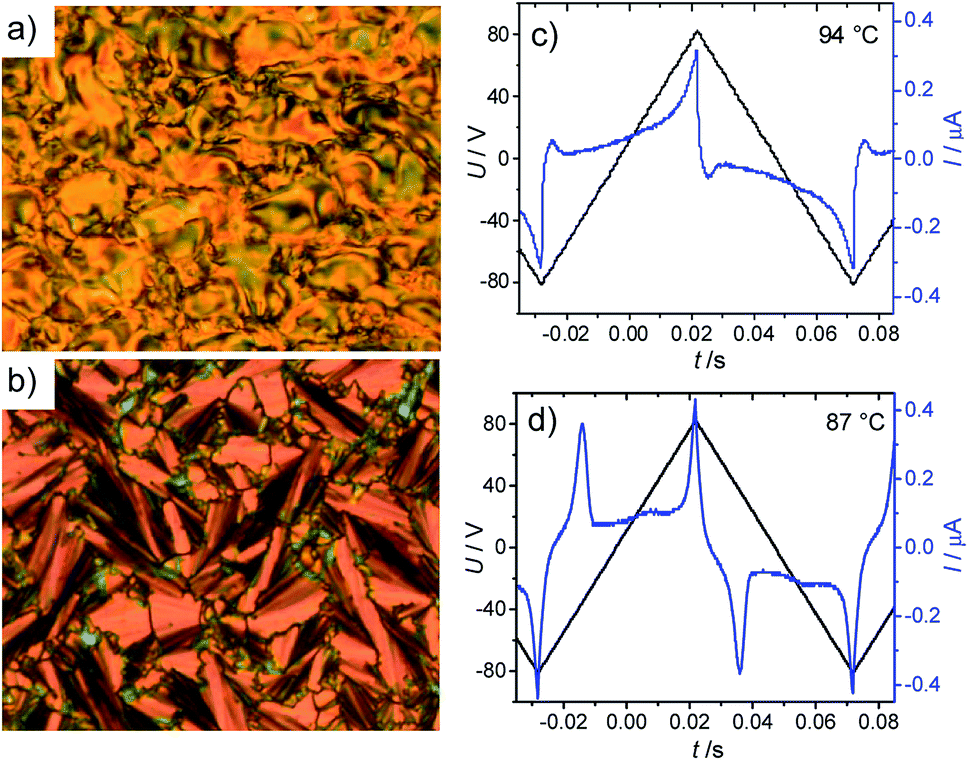
The optically isotropic DC[*] phase is completely removed for the longest derivative in the series BrF2n with n = 16. On cooling BrF216 from the isotropic liquid state a birefringent schlieren texture is observed below T = 96 °C in a homeotropic cell (Fig. 5a) and a fan texture is observed in a planar cell where the dark extinctions are inclined by an angle of about 28°, indicating a synclinic tilted smectic phase (SmCs phase, see Fig. 5b). XRD investigation of this phase was not possible due to the rapid crystallization of the monotropic phase. In electro-optical investigations two polarization current peaks in each half period of an applied triangular wave voltage were observed (Fig. 5c and d). The two polarization peaks are weak and widely separated at the Iso–SmCs transition, growing in intensity and coming a bit closer to each other on further cooling, reaching a polarization values P = 290 nC cm−2. The mode of appearance of the polarization peaks, their shape and the relatively small polarization values are typical for SmCsPAR phases with randomized polar order, described in detail for 4-cyanoresorciol based BCLCs.51 Therefore, the LC phase of compound BrF216 is assigned as SmCsPAR phase. In this smectic phase ferroelectric domains with synclinic and antipolar correlation form a field induced SmCsPF state which relaxes at reduced voltage back to a macroscopically antipolar structure. The position of the dark extinctions does not change, indicating the relaxation and switching take place by rotation around the molecular long axis (see Fig. S11†).
 |
| | Fig. 5 Textures and polarization current response curve of the SmCsPAR phase of compound BrF216: textures as observed at T = 93 °C (a) in a homeotropic cell; (b) in a 6 μm coated ITO cell with planar alignment; (c) and (d) switching current response curves in the same ITO cell as recorded under a triangular wave voltage 160 Vpp (10 Hz, 5 kΩ) at the indicated temperatures. | |
3.3. Investigation of mixtures of compounds BrF2n with 5-CB
It is well known that the two different types of soft crystalline DC[*] phases (HNF and HNC) behave differently upon mixing with 4′-n-pentyl-4-cyanobiphenyl (5-CB). The chirality of HNF phases can be retained even at high dilution with a nematic LC host (>95%),53 while the HNC phases can be diluted only by a small amount (to ∼50%) of a nematic LC (5-CB) without loss of the DC[*] phase and chirality.45–47 The mixtures of selected compounds BrF2n with 5-CB were investigated and compared with the results obtained for their related monofluorinated analogues BrFn (see Table 2).
Table 2 Phase transition temperatures and mesophase types of 1:1 mixtures of 5-CB and compounds BrF10–BrF14 and comparison with related 4-bromoresorcinol derivatives BrF10–BrF14a46
| Mixture |
Heating T/°C |
Cooling T/°C |
|
Transition temperatures were taken from the observed textures using the polarized optical microscopy; abbreviations: N = nematic phase; Cr[*] = crystalline phase composed of a conglomerate of chiral domains; for other abbreviations please see Table 1.
|
|
BrF210 + 5-CB |
Cr 68 Iso |
Iso 47 N 39 Cr |
|
BrF212 + 5-CB |
DC[*] 60 Iso |
Iso 56 DC[*] |
|
BrF214 + 5-CB |
DC[*] 58 Iso |
Iso 54 DC[*] |
|
BrF10 + 5-CB |
Cr 54 Iso |
Iso 42 N 33 Cr |
|
BrF12 + 5-CB |
Cr[*] 40 DC[*] 55 Iso |
Iso 53 DC[*] 38 Cr[*] |
|
BrF14 + 5-CB |
Cr 38 DC[*] 64 Iso |
Iso 46 DC[*] 35 Cr |
The DC[*] phase of the pure compound BrF210 is removed in its 1:1 mixture with 5CB and a direct transition from the crystalline state to the isotropic liquid takes place at 68 °C on heating (Table 2). On cooling the same mixture from the isotropic liquid state a monotropic nematic phase is formed which crystallizes at T ∼39 °C without the formation of DC[*] phase. The next two homologues BrF212 and BrF214 behave differently; DC[*] phases are formed in their 1:1 mixtures with 5CB as room temperature mesophases and no crystallization takes place either on heating or cooling (see Fig. 6). Similar to the other HNC phases, only a limited amount of 5-CB can be mixed into the HNC phase of BrF2n and any further increase of the amount of 5-CB removes the DC[*] phases. Comparing the results obtained for BrF2n compounds with their monofluorinated analogues (compounds BrFn, see Table 2)47 indicate that both types of compounds behave similarly in their mixed systems, which further confirms the similarity of the HNC phases exhibited by the azobenzene based bent-core mesogens.
 |
| | Fig. 6 Textures of the DC[*] phase of 1:1 mixture of compound BrF214 with 5CB at T = 45 °C: (a) after rotating one polarizer by 10° from the crossed position in clock-wise direction and (b) in anticlockwise direction, showing dark and bright domains, indicating the presence of areas with opposite chirality sense. | |
3.4. Compounds ClF2n with chlorine at the apex
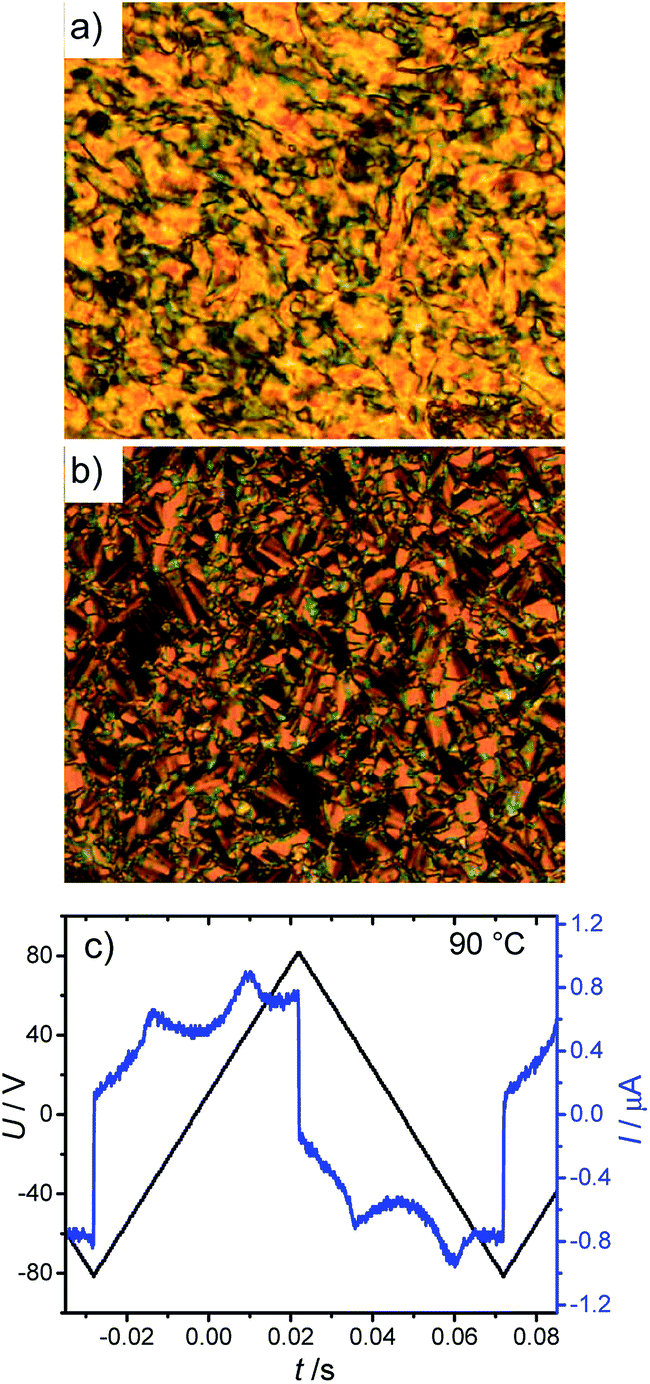
The effect of replacing bromine by a smaller chlorine on the mesophase type has been investigated with the 4-chlororesorcinol derived compounds ClF2n (see Table 3 for data and Fig. 7 for DSC traces for ClF214). Unlike series BrF2n, compounds ClF2n do not show any DC[*] phase; instead they form monotropic smectic C phases. On cooling the shorter homologues with n = 8 and 10 from the isotropic liquid state a birefringent SmC phase is observed at the same temperature T = 86 °C for both derivatives. The investigations of these SmC phases was not possible due to the rapid crystallization starting directly after the appearance of the SmC and for the same reason the value of transition enthalpy for the Iso–SmC transition cannot be separated from that of SmC–Cr transition. For the next homologues with n ≥ 12 the SmC phases are formed in a sufficient temperature range enabling electro-optical investigations. The temperature range of the SmC phases in the series ClF212–ClF216 is increasing with increasing the chain length (see Table 3). Fig. 8a and b shows the textures observed for the SmC phase of compound ClF216 upon cooling from the isotropic liquid state, where a birefringent schlieren texture is observed in a homeotropic cell (Fig. 8a). A broken fan texture with the dark extinctions inclined by an angle of 26°, indicating a synclinic tilted smectic phase (SmCs phase) is observed in a planar cell (Fig. 8b).
Table 3 Phase transition temperatures (T/°C), mesophase types, and transition enthalpies [ΔH/kJ mol−1] of compounds ClF2na
|

|
| Compound |
n
|
Heating T/°C [ΔH/kJ mol−1] |
Cooling T/°C [ΔH/kJ mol−1] |
|
The phase transition temperatures were measured as mentioned in Table 1.
The value of Iso–SmC transition enthalpy cannot be separated from SmC–Cr transition value. Abbreviations: SmCx = smectic C phase with unknown polar structure, most likely also representing SmCsPAR phases; for other abbreviations see Table 1.
|
|
ClF28
|
8 |
Cr 98 [33.1] Iso |
Iso 86 [30.7] Cr + SmCxb |
|
ClF210
|
10 |
Cr 101 [35.9] Iso |
Iso 86 [35.7] Cr + SmCxb |
|
ClF212
|
12 |
Cr 101 [25.7] Iso |
Iso 94 [6.3] SmCsPAR 80 [18.5] Cr |
|
ClF214
|
14 |
Cr 101 [37.9] Iso |
Iso 98 [8.6] SmCsPAR 83 [27.5] Cr |
|
ClF216
|
16 |
Cr 105 [35.7] Iso |
Iso 100 [7.5] SmCsPAR 75 [37.2] Cr |
 |
| | Fig. 7 DSC heating and cooling scans of ClF214 with a rate of 10 K min−1. | |
 |
| | Fig. 8 Textures and polarization current response curve of the SmCsPAR phase of compound ClF216 at T = 90 °C: (a) texture as observed in a homeotropic cell; (b) texture observed in a 6 μm coated ITO cell with planar alignment and (c) switching current response curve in the same ITO cell as recorded under a triangular wave voltage 160 Vpp (10 Hz, 5 kΩ). | |
Under an applied triangular wave voltage of 160 Vpp in a 6 μm ITO cell two broad polarization peaks per half period of the applied voltage appears in the SmC phase of all investigated ClF2n compounds with n = 12–16 with a polarization value P ∼ 100 nC cm−2 (see Fig. 8c for ClF216). Based on the textures and the shape of the polarization curves, the SmC phases exhibited by these materials are assigned as SmCsPAR phases, but the shape and the polarization values are very distinct from that of the analogous bromine substituted compound BrF216. The broad diffuse shape of the polarization current peaks is similar to that observed for previously reported SmAPAR phase showing Langevin-type switching.54 Therefore, we conclude that the switching of the chlorine substituted compounds is also typically Langevin type,55 meaning that small polar domains grow under the electric field and then can be switched between two polar states. This means that the polar domains in the SmC phase of ClF216 might have a broader size distribution than those observed in the SmCsPAR phase of BrF216 and grow to larger domains under the applied field. There is a larger threshold voltage for polar switching of BrF216 compared to ClF216 though the magnitude of polarization is higher (280 vs. 100 nC cm−2). Probably there is larger polarization in the ferroelectric domains of BrF216 but also a stronger antipolar correlation between these domains. It appears that bromine substitution favours polar packing compared to the smaller chlorine, though it could be expected that the bend of the bromine substituted compound might be a bit smaller than that of the chlorine substituted,50a and additionally, the bulkier bromine is expected to reduce the core packing density for steric reasons. This effect might possibly be attributed to the higher polarizability of bromine and the C–Br bond.
3.5. Comparison with related compounds
Table 4 shows a comparison between azobenzene derived BCLCs with 4-bromoresorcinol and 4-chlororesorcinol cores and n = 12 chain length, but with different number of peripheral fluorine substituents. Before discussing the effect of fluorine substitution, it is mentioned here that the larger bromine atom (cv ∼ 33 nm3, crystal volumes cv of Immirzi)56 in the 4-position at the resorcinol core favours the formation of HNC phase in all cases compared to the smaller chlorine (cv ∼ 27 nm3),56 indicating that the size of the substituent at the apex and the degree of molecular twist induced by this substituent are important for layer distortion and chirality synchronization required for DC[*] phase formation.47 For the BCLCs with 4-bromoresorcinol central core, peripheral fluorination removes the nematic phase and stabilizes the HNC phase, most probably by increasing the attractive π–π interactions by reduction of the electron density of the aromatics. However, inserting an additional fluorine atom in the meta position with respect to the terminal alkyl chain (compound BrF212) reduces the HNC phase stability (reduced transition temperature Iso-DC) and favours crystallization. Reduction of HNC phase stability is probably due to the steric effect of the additional fluorine, reducing the packing density. The increased melting points are attributed to the improved π–π-stacking ability caused by the further reduction of the electron density by the additional electron withdrawing fluorine.
Table 4 Phase transition temperatures (T/°C) and mesophase types for different types of 4-bromoresorcinol and 4-chlororesorcinol derived BCLCs with azobenzene wings and the effect of peripheral F-atoms (Y, Y′ = H, F)a
|

|
| Comp. |
X |
Y |
Y′ |
T/°C |
Ref. |
|
Abbreviations: N = nematic phase; SmCaPA = anticlinic antiferrelectric switching SmC phase (B2 phase); for other abbreviations see Table 1.
|
|
Br12
|
Br |
H |
H |
DC[*] 93 (N 83) Iso |
49
|
|
BrF12
|
Br |
F |
H |
DC[*] 106 Iso |
47
|
|
BrF212
|
Br |
F |
F |
Cr1 97 Cr2 103 (DC[*] 90) Iso |
|
|
Cl12
|
Cl |
H |
H |
Cr1 90 Cr2 103 (N 97) Iso |
49
|
|
ClF12
|
Cl |
F |
H |
Cr 115 (SmCaPA 97) Iso |
47
|
|
ClF212
|
Cl |
F |
F |
Cr 101 (SmCsPAR 97) Iso |
|
In the case of the compounds derived from 4-chlororesorcinol the core fluorination removes the nematic phase, too, but in this case it leads first to a macroscopically polar (SmCaPA) and then, after addition of the next fluorine, to a locally polar tilted smectic phase (SmCsPAR, see Table 4). The mesophase stability of the LC phases is apparently not affected by fluorination, but the introduction of the second fluorine in m-position removes the long range polar order achieved for the monofluorinated compound (two sharp polarization peaks, Ps ∼ 500 nC cm−2, see Fig. S11† in ref. 47) and replaces this by a local polar domain structure (two diffuse polarization peaks, Ps ∼ 100 nC cm−2, Fig. 8c). Simultaneously the additional F substituents change the tilt correlation in the smectic phase from anticlinic in ClF12 to synclinic in ClF212 and the switching process from rotation on the tilt-cone for ClF12 (Fig. S10† in ref. 47) to a reorganization around the long axis in the case of ClF2n (Fig. S11†). This is most probably an effect of the reduced packing density, favoured by the increased bulkiness of the rod-like wings with two adjacent fluorines in each azobenzene wing. Overall, there seems to be a delicate balance of the influence of the two competing effects. It appears that the packing density is increased most efficiently by the electron accepting 3-fluorination adjacent to the 4-alkyloxy chain, whereas 2-fluorination in meta position to the alkyloxy chain contributes more to the unfavourable steric effect of fluorine, thus reducing the packing density. Besides the steric and electronic effects of fluorination, fluorine substitution can also have an effect on the conformation of the Ar–O–CH2 linkage by influencing orbital interactions57 and by strengthening weak intra-and intermolecular hydrogen bonding involving C–H bonds,58 thus supporting the twisting of the molecules and the formation of DC[*] phases.
4. Summary and conclusions
Two series of new bent-core liquid crystalline materials combining 4-bromoresorcinol or 4-chlororesorcinol cores with two 2,3-difluorinated and 4-alkoxy substituted azobenzene side arms have been synthesized and investigated. Depending on the size of the halogen atom in 4-position of the central bent core unit (Cl vs. Br) and on the length of the terminal alkyl chains different types of mesophases were observed. It was found that the majority of 4-bromoresorcinol derived compounds (n = 10–14) form HNC-type DC[*] phases which are replaced by a SmCsPAR phase upon chain elongation (n = 16). For the 4-chlororesorcinol derivatives only SmC phases, but no DC[*] phases were observed. The investigated SmC phases represent SmCsPAR type polar domain phases. The SmCsPAR phase of the chlorine substituted compound is distinct from that of the related bromine derivative by the significantly broader polarization peaks and the smaller polarization values, indicating a Langevin-type switching (field-induced growth of the polar domains) for the chlorinated compound and a more superparaelectric type of switching59 (fusion of already existing polar domains) for the brominated compound. The DC[*] phases exhibited by these materials represent helical nano-crystallites phases (HNC) but with a different local structure if compared with the HNC phases of the related azobenzene compound without peripheral fluorine or with only one fluorine.47,49 Though, core fluorination can favour HNC phase formation and modifies the precise phase structure, it cannot induce DC phases if the core unit would not also support its formation. It also cannot fundamentally change the structure of the DC[*] phase to HNF or fluid DC phases. Introduction of the first fluorine adjacent to the alkoxy chains obviously favors layer formation and DC[*] phase formation (removal of N phases) and development of polar order, most probably by increasing the attractive π–π interactions by reduction of the electron density of the aromatics. The second fluorine appears to reduce the DC[*] phase stability a bit and appears to reduce the coherence length of local polarization, probably due to its steric effect, reducing the packing density a bit. Thus fluorination is a tool for tailoring HNC phase ranges and the fine structure of the HNC phases. Future work will be devoted to a more detailed analysis of the HNC phases by imaging methods and the investigation of the effects of photoisomerization of the azobenzene units incorporated in the molecular structure of these BCLCs by polarized and nonpolarized light. This could lead to additional possibilities for phase modulation and chirality modulation, which could result in potentially useful applications.
Acknowledgements
The work was supported by the DFG (Grant Ts 39/24-1). M. Alaasar is grateful to the Alexander von Humboldt Foundation for the research fellowship at the Martin-Luther University Halle-Wittenberg, Germany.
References
- T. Niori, T. Sekine, J. Watanabe, T. Furukawa and H. Takezoe, J. Mater. Chem., 1996, 6, 1231–1233 RSC.
-
(a) R. A. Reddy and C. Tschierske, J. Mater. Chem., 2006, 16, 907–961 RSC;
(b) H. Takezoe and Y. Takanishi, Jpn. J. Appl. Phys., Part 1, 2006, 45, 597–625 CrossRef CAS;
(c) A. Eremin and A. Jákli, Soft Matter, 2013, 9, 615–637 RSC;
(d) J. Etxebarria and M. B. Ros, J. Mater. Chem., 2008, 18, 2919–2926 RSC;
(e) M. Alaasar, Liq. Cryst., 2016 DOI:10.1080/02678292.2016.1175676.
-
C. Tschierske, Nanoscale stereochemistry in liquid crystals, in Chirality at the nanoscale, ed. D. B. Amabilino, Wiley-VCH, Weinheim, 2009, pp. 271–304 Search PubMed.
- H. Takezoe, Top. Curr. Chem., 2012, 318, 303–330 CrossRef CAS PubMed.
- C. Tschierske, Angew. Chem., Int. Ed., 2013, 52, 8828–8878 CrossRef CAS PubMed.
- C. Tschierske and G. Ungar, ChemPhysChem, 2016, 17, 9–26 CrossRef CAS PubMed.
- K.-U. Jeong, B. S. Knapp, J. J. Ge, S. Jin, M. J. Graham, F. W. Harris and S. Z. D. Cheng, Chem. Mater., 2006, 18, 680–690 CrossRef CAS.
- C. Roche, H.-J. Sun, M. E. Prendergast, P. Leowanawat, B. E. Partridge, P. A. Heiney, F. Araoka, R. Graf, H. W. Spiess, X. B. Zeng, G. Ungar and V. Percec, J. Am. Chem. Soc., 2014, 136, 7169–7185 CrossRef CAS PubMed.
-
(a) V. P. Panov, M. Nagaraj, J. K. Vij, Y. P. Panarin, A. Kohlmeier, M. G. Tamba, R. A. Lewis and G. H. Mehl, Phys. Rev. Lett., 2010, 105, 167801 CrossRef CAS PubMed;
(b) V. Borshch, Y.-K. Kim, J. Xiang, M. Gao, A. Jakli, V. P. Panov, J. K. Vij, C. T. Imrie, M. G. Tamba, G. H. Mehl and O. D. Lavrentovich, Nat. Commun., 2013, 4, 2635 CAS.
- M. Cestari, S. Diez-Berart, D. A. Dunmur, A. Ferrarini, M. R. de la Fuente, D. J. B. Jackson, D. O. Lopez, G. R. Luckhurst, M. A. Perez-Jubindo, R. M. Richardson, J. Salud, B. A. Timimi and H. Zimmermann, Phys. Rev. E, 2011, 84, 031704 CrossRef CAS PubMed.
- D. Chen, J. H. Porada, J. B. Hooper, A. Klittnick, Y. Shen, M. R. Tuchband, E. Korblova, D. Bedrov, D. M. Walba, M. A. Glaser, J. E. Maclennan and N. A. Clark, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 15931–15936 CrossRef CAS PubMed.
- S.-W. Choi, T. Izumi, Y. Hoshino, Y. Takanishi, K. Ishikawa, J. Watanabe and H. Takezoe, Angew. Chem., Int. Ed., 2006, 45, 1382–1385 CrossRef CAS PubMed.
-
H. –S. Kitzerow and C. Bahr, Chirality in Liquid Crystals, Springer-Verlag, New York, 2001 Search PubMed.
- A. Belaissaoui, S. J. Kowling and J. W. Goodby, Liq. Cryst., 2013, 40, 822–830 CrossRef CAS.
- D. Chen, H. Wang, M. Li, M. A. Glaser, J. E. Maclennan and N. A. Clark, Soft Matter, 2014, 10, 9105–9109 RSC.
- L. E. Hough, M. Spannuth, M. Nakata, D. A. Coleman, C. D. Jones, G. Dantlgraber, C. Tschierske, J. Watanabe, E. Körblova, D. M. Walba, J. E. Maclennan, M. A. Glaser and N. A. Clark, Science, 2009, 325, 452–456 CrossRef CAS PubMed.
- L. E. Hough, H. T. Jung, D. Krüerke, M. S. Heberling, M. Nakata, C. D. Jones, D. Chen, D. R. Link, J. Zasadzinski, G. Heppke, J. P. Rabe, W. Stocker, E. Körblova, D. M. Walba, M. A. Glaser and N. A. Clark, Science, 2009, 325, 456–460 CrossRef CAS PubMed.
- D. Chen, R. Shao, J. E. Maclennan, M. A. Glaser, E. Körblova, D. M. Walba, N. Gimeno, M. B. Ros and N. A. Clark, Liq. Cryst., 2013, 40, 1730–1735 CrossRef CAS.
-
(a) I. Dierking, Angew. Chem., Int. Ed., 2010, 49, 29–30 CrossRef CAS PubMed;
(b) J. P. F. Lagerwall and F. Giesselmann, ChemPhysChem, 2010, 11, 975–977 CrossRef CAS PubMed.
- V. Görtz, Liq. Cryst. Today, 2010, 19, 37–48 CrossRef.
-
(a) M. Alaasar, M. Prehm, M. Nagaraj, J. K. Vij and C. Tschierske, Adv. Mater., 2013, 25, 2186–2191 CrossRef CAS PubMed;
(b) M. Alaasar, M. Prehm, K. May, A. Eremin and C. Tschierske, Adv. Funct. Mater., 2014, 24, 1703–1717 CrossRef CAS.
- C. Dressel, F. Liu, M. Prehm, X.-B. Zeng, G. Ungar and C. Tschierske, Angew. Chem., Int. Ed., 2014, 53, 13115–13120 CrossRef CAS PubMed.
- C. Dressel, T. Reppe, M. Prehm, M. Brautzsch and C. Tschierske, Nat. Chem., 2014, 6, 971–977 CrossRef CAS PubMed.
- M. Alaasar, M. Prehm, Y. Cao, F. Liu and C. Tschierske, Angew. Chem., Int. Ed., 2016, 55, 312–316 CrossRef CAS PubMed.
- A. Yoshizawa, Y. Kato, H. Sasaki, Y. Takanishi and J. Yamamoto, J. Phys. Chem. B, 2016, 120, 4843–4851 CrossRef CAS PubMed.
-
(a) G. Dantlgraber, A. Eremin, S. Diele, A. Hauser, H. Kresse, G. Pelzl and C. Tschierske, Angew. Chem., Int. Ed., 2002, 41, 2408–2414 CrossRef CAS;
(b) C. Keith, R. A. Reddy, A. Hauser, U. Baumeister and C. Tschierske, J. Am. Chem. Soc., 2006, 128, 3051–3066 CrossRef CAS PubMed.
- G. Heppke, D. D. Parghi and H. Sawade, Liq. Cryst., 2000, 27, 313–320 CrossRef CAS.
- J. Thisayukta, Y. Nakayama, S. Kawauchi, H. Takezoe and J. Watanabe, J. Am. Chem. Soc., 2000, 122, 7441–7448 CrossRef CAS.
- R. A. Reddy and B. K. Sadashiva, Liq. Cryst., 2003, 30, 1031–1050 CrossRef.
- A. Roy, M. Gupta, S. Radhika, B. K. Sadashiva and R. Pratibha, Soft Matter, 2012, 8, 7207–7214 RSC.
- J. Ortega, C. L. Folcia, J. Etxebarria, N. Gimeno and M. B. Ros, Phys. Rev. E: Stat., Nonlinear, Soft Matter Phys., 2003, 68, 11707 CrossRef CAS PubMed.
- S. Kang, Y. Saito, N. Watanabe, M. Tokita, Y. Takanishi, H. Takezoe and J. Watanabe, J. Chem. Phys. B, 2006, 110, 5205–5214 CrossRef CAS PubMed.
-
(a) M. Nagaraj, K. Usami, Z. Zhang, V. Görtz, J. W. Goodby and H. F. Gleeson, Liq. Cryst., 2014, 41, 800–811 CrossRef CAS;
(b) M. Nagaraj, J. C. Jones, V. P. Panov, H. Liu, G. Portale, W. Bras and H. F. Gleeson, Phys. Rev. E, 2015, 91, 042504 CrossRef CAS PubMed.
-
(a) S. K. Lee, X. Li, S. Kang, M. Tokitra and J. Watanabe, J. Mater. Chem., 2009, 19, 4517–4522 RSC;
(b) T. Bao, K. Wang and M.-S. Zhan, Liq. Cryst., 2014, 41, 1687–1695 CrossRef CAS.
-
(a) A. Jákli, Y.-M. Huang, K. Fodor-Csorba, A. Vajda, G. Galli, S. Diele and G. Pelzl, Adv. Mater., 2003, 15, 1606–1610 CrossRef;
(b) W. Weissflog, M. W. Schröder, S. Diele and G. Pelzl, Adv. Mater., 2003, 15, 630–633 CrossRef CAS.
-
(a) S. K. Lee, L. Shi, M. Tokita, H. Takezoe and J. Watanabe, J. Phys. Chem. B, 2007, 111, 8698–8701 CrossRef CAS PubMed;
(b) S. K. Lee, L. Shi, M. Tokita and J. Watanabe, J. Phys. Chem. B, 2008, 112, 6762–6766 CrossRef CAS PubMed.
-
(a) D. M. Walba, L. Eshat, E. Körblova and R. K. Shoemaker, Cryst. Growth Des., 2005, 5, 2091–2099 CrossRef CAS;
(b) D. Chen, J. E. Maclennan, R. Shao, D. K. Yoon, H. Wang, E. Körblova, D. M. Walba, M. A. Glaser and N. A. Clark, J. Am. Chem. Soc., 2011, 133, 12656–12663 CrossRef CAS PubMed.
- J. M. -Perdiguero, I. Alonso, C. L. Folcia, J. Etxebarria and J. Ortega, J. Mater. Chem., 2009, 19, 5161–5166 RSC.
- C. Zhang, N. Diorio, O. D. Lavrentovich and A. Jákli, Nat. Commun., 2014, 5, 3302 CAS.
-
(a) J. Thisayukta, H. Takezoe and J. Watanabe, Jpn. J. Appl. Phys., Part 1, 2001, 40, 3277 CrossRef CAS;
(b) H. Niwano, M. Nakata, J. Thisayukta, D. R. Link, H. Takezoe and J. Watanabe, J. Phys. Chem. B, 2004, 108, 14889–14896 CrossRef CAS;
(c) H. Kurosu, M. Kawasaki, M. Hirose, M. Yamada, S. Kang, J. Thisayukta, M. Sone, H. Takezoe and J. Watanabe, J. Phys. Chem. A, 2004, 108, 4674–4678 CrossRef CAS.
- H. Kresse, J. Saltetnokova, H. Nadasi, W. Weissflog and A. Hauser, Liq. Cryst., 2001, 28, 1017–1023 CrossRef CAS.
-
(a) E. Bialecka-Florjanczyk, I. Sledzinska, E. Górecka and J. Przedmojski, Liq. Cryst., 2008, 35, 401–406 CrossRef CAS;
(b) A. Zep, M. Salamonczyk, N. Vaupotič, D. Pociecha and E. Gorecka, Chem. Commun., 2013, 49, 3119–3121 RSC.
- T. Niori, T. Sekine, J. Watanabe, T. Furukawa and H. Takezoe, J. Mater. Chem., 1996, 6, 1231–1233 RSC.
- C. Zhu, C. Wang, A. Young, F. Liu, I. Gunkel, D. Chen, D. Walba, J. Maclennan, N. Clark and A. Hexemer, Nano Lett., 2015, 15, 3420–3424 CrossRef CAS PubMed.
- M. Alaasar, M. Prehm and C. Tschierske, Chem. Commun., 2013, 49, 11062–11064 RSC.
- M. Alaasar, M. Prehm, M. Brautzsch and C. Tschierske, J. Mater. Chem. C, 2014, 2, 5487–5501 RSC.
- M. Alaasar, M. Prehm, M. Brautzsch and C. Tschierske, Soft Matter, 2014, 10, 7285–7296 RSC.
- M. Alaasar, M. Prehm and C. Tschierske, Chem.–Eur. J., 2016, 22, 6583–6597 CrossRef CAS PubMed.
- M. Alaasar, M. Prehm and C. Tschierske, Liq. Cryst., 2013, 40, 656–668 CrossRef CAS.
- For a comparison of chlorine and bromine substituted BCLCs with phenylbenzoate wings, see:
(a) W. Weissflog, S. Sokolowski, H. Dehne, B. Das, S. Grande, M. W. Schröder, A. Eremin, S. Diele, G. Pelzl and H. Kresse, Liq. Cryst., 2004, 31, 923–933 CrossRef CAS;
(b) S. Kang, J. Thisayukta, H. Takezoe, J. Watanabe, K. Ogino, T. Doi and T. Takahashi, Liq. Cryst., 2004, 31, 1323–1336 CrossRef CAS.
- M. Alaasar, M. Prehm, M.-G. Tamba, N. Sebastian, A. Eremin and C. Tschierske, ChemPhysChem, 2016, 17, 278–287 CrossRef CAS PubMed.
- K. Miyasato, S. Abe, H. Takezoe, A. Fukuda and E. Kuze, Jpn. J. Appl. Phys., Part 1, 1983, 22, L661–L663 CrossRef.
-
(a) T. Otani, F. Araoka, K. Ishikawa and H. Takezoe, J. Am. Chem. Soc., 2009, 131, 12368 CrossRef CAS PubMed;
(b) F. Araoka, G. Sugiyama, K. Ishikawa and H. Takezoe, Opt. Mater. Express, 2011, 1, 27 CrossRef CAS.
- K. Gomola, L. Guo, D. Pociecha, F. Araoka, K. Ishikawa and H. Takezoe, J. Mater. Chem., 2010, 20, 7944–7952 RSC.
- Y. Shimbo, E. Gorecka, D. Pociecha, F. Araoka, M. Goto, Y. Takanishi, K. Ishikawa, J. Mieczkowski, K. Gomola and H. Takezoe, Phys. Rev. Lett., 2006, 97, 113901 CrossRef PubMed.
- A. Immirzi and B. Perini, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1977, 33, 216–218 CrossRef.
-
A. J. Kirby, The anomeric effect and related stereoelectronic effects at oxygen, Springer, Berlin, Heidelberg, 1983 Search PubMed.
-
(a) T. Steiner and G. R. Desiraju, Chem. Commun., 1998, 8, 891–892 RSC;
(b) O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed.
- M. F. Achard, J. P. Bedel, J. P. Marcerou, H. T. Nguyen and J. C. Rouillon, Eur. Phys. J. E, 2003, 10, 129–134 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra18482k |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab,
Marko
Prehm
a and
Carsten
Tschierske
*a
*ab,
Marko
Prehm
a and
Carsten
Tschierske
*a