
Creating hierarchically macro-/mesoporous Sn/CeO2 for the selective catalytic reduction of NO with NH3†
Cheng Fang,
Liyi Shi,
Hongrui Li,
Lei Huang,
Jianping Zhang and
Dengsong Zhang*
Research Center of Nano Science and Technology, Shanghai University, Shanghai 200444, China. E-mail: dszhang@shu.edu.cn
First published on 11th August 2016
Abstract
A hierarchically macro-/mesoporous Sn/CeO2 (H-Sn/CeO2) catalyst with uniform element composition and dispersion was successfully synthesized by using KIT-6 as a hard template for the selective catalytic reduction (SCR) of NO with NH3. This catalyst was mainly characterized by such techniques as transmission electron microscopy, scanning electron microscopy, X-ray diffraction, laser Raman spectroscopy, N2 adsorption–desorption analysis, X-ray photoelectron spectroscopy, H2 temperature-programmed reduction and NH3 temperature-programmed desorption. The obtained catalyst possesses hierarchical nanostructures with large specific surface area and good dispersion of elements. It was found that the hierarchically nanostructured Sn/CeO2 catalyst exhibited high NH3-SCR activity and a broad operating temperature window. It has been demonstrated that the Sn atoms incorporate into the crystal lattice of ceria, and more Ce3+ and Oα species exist on the surface of H-Sn/CeO2. It is interesting that the doping of Sn element could improve the reducibility of the H-Sn/CeO2 catalyst due to the synergetic interaction between Ce and Sn species. In addition, the H-Sn/CeO2 catalyst possesses more acidic sites and stronger acidic strength. Therefore, the obtained catalyst exhibits excellent stability and water resistance as well as enhanced SO2 tolerance.
1. Introduction
Generally, nitrogen oxides (NOx) emitted from the combustion of fossil fuels can give rise to a variety of environmental problems, including acid rain, photochemical smog, city haze weather and the greenhouse effect.1–3 Moreover, they are harmful to human health and cause some diseases, such as bronchitis, emphysema and even cancer. In terms of the many technologies for eliminating NOx, selective catalytic reduction of NOx with NH3 (NH3-SCR) is considered to be one of the most efficient and widely used methods.4–7 A series of vanadium-based catalysts are used worldwide in industry for removing NOx in a temperature range of 300–400 °C.8–10 However, these highly effective catalysts suffer from several inevitable drawbacks, for instance, SO2 oxidation to SO3, release of toxic vanadium species to the environment at high temperatures and especially poor activity in low-temperature regions.11,12 Therefore, it is extremely desirable to develop alternative deNOx catalysts to overcome these issues.Nowadays, several transition metal oxides and rare earth metal catalysts have been investigated owing to their ready synthesis and good redox properties.13–16 Among them, ceria has attracted considerable attention for its excellent oxygen storage capacity and redox properties via the shift between Ce4+ and Ce3+ under oxidizing and reducing conditions, respectively.17,18 It is noted that pure CeO2 expresses unsatisfactory NH3-SCR activity, but its performance can be significantly improved when it is modified by other metal oxides.19–21 In recent years, a series of Ce-based catalysts have been reported such as MnOx–CeO2,22 CeO2–WO3,20 WO3/CeO2–ZrO2,23 CeO2/TiO2,21,24,25 CeO2/Al2O3,26 CeO2 modified MnOx/TiO2,27 all of these catalysts have exhibited good catalytic performance and N2 selectivity. However, the residual SO2 in the flue gas could poison the catalysts and restrict the catalytic activity in practical applications.28 Thus there is still a lot of room to improve the SO2 tolerance of Ce-based catalysts.29,30
SnO2 is a very important semiconductor and has a large number of intrinsic defects in its structure, which has been extensively applied in gas sensors, battery technology and catalysis.31,32 It has been reported that SnO2 brings about a strong Lewis acid and the NH3 can favourably chemically adsorb on the surface of SnO2 to form a NH3–Sn bond.33 Park and Liu et al. found that Sn4+ was active for the reduction of NOx.34,35 Chang et al. reported that the introduction of SnO2 could improve the low temperature NH3-SCR activity and SO2 resistance of MnOx–CeO2.36 Li et al. also found that Ce–Sn–Ox catalysts exhibited good NH3-SCR activity in a broad temperature range and showed high resistance to H2O and SO2.37 Nevertheless, the use of Sn precursor must be raised in order to get higher NOx conversion, which will not only increase costs but also be harmful to human health and environment. Besides, these Ce–Sn–Ox catalysts were prepared by a facile co-precipitation method, which makes it difficult to ensure that active components are uniformly dispersed and thus the aggregates are prone to form during the catalytic process, resulting in the deactivation of catalysts.38 Therefore, it is worthwhile to develop high-performance Ce–Sn–Ox catalysts with stable and uniformly distributed active components on the surface.
It is well established that microlevel control of the structure, size, and shape of inorganic materials can lead to novel catalytic properties that are not found in corresponding bulk materials.39,40 Recently, we found that the hollow porous MnxCo3−xO4 nanocages synthesized via a self-assemble method exhibit a much better catalytic performance than conventional MnxCo3−xO4 nanoparticles and the main reason can be ascribed to the hollow and porous structures.14 Nowadays, hierarchically macro-/mesoporous materials with high specific surface area and abundant porous structure have aroused extensive attention, because they may possess numerous edges and corners for the adsorption and activation of reactants and the interconnected pores are conducive to the reactant molecular diffusion.41 Compared to the conventional nanoparticles, the meso-pores in the hierarchical nanostructure are particularly important for metal oxides. It is noted that mesoporous metal oxides are of much interest for energy conversion and storage, magnetic devices and catalysis.42,43 Huang et al. reported that Fe–Mn oxide could be dispersed well on the mesoporous SiO2 support.44 Yu et al. found that the mesoporous structure could improve the SO2-resistance of the MnO2–Fe2O3–CeO2–TiO2 catalyst.45 However, to the best of our knowledge, hierarchically macro-/mesoporous Sn/CeO2 catalyst has not yet been reported for SCR of NO with NH3.
In this paper, hierarchically macro-/mesoporous Sn/CeO2 catalyst was synthesized by using KIT-6 as hard template for selective catalytic reduction of NO with NH3. The schematic illustration of this unique catalyst is shown in Fig. 1. For comparison, the corresponding usual Sn/CeO2 catalysts without special pore structure were also prepared by common method and discussed under the same working conditions. These catalysts were characterized systematically, and their NH3-SCR activity, stability, SO2 tolerance and H2O resistance were also investigated.
 | ||
| Fig. 1 Schematic illustration of the synthetic procedure of the hierarchically macro-/mesoporous catalysts with KIT-6. | ||
2. Experimental section
2.1 Catalyst preparation
All the chemicals were purchased from Sinopharm Chemical Regent Company and used without further purification. The template KIT-6 was synthesized according to the previous literature (hydrothermal treatment at 100 °C).46In a typical synthesis process, 2.93 g of Ce(NO3)3·6H2O, 0.26 g of SnCl4·5H2O and 0.25 g of KNO3 were dissolved in 2.5 mL of deionized water. A certain amount of 0.8 M ammonia solution was added into the above solution and stirred for several minutes to form solution A. Secondly, 2.0 g KIT-6 was suspended in 80 mL n-hexane and stirred at room temperature for 3 h. Then, 2.0 mL solution A was added dropwise with stirring. The mixture was stirred overnight, filtered and dried completely at room temperature. Subsequently, the mixture was calcined in a muffle furnace at 500 °C (1 °C min−1) for 5 h with a cover in a normal crucible. Finally, the sample was suspended in a 2 M NaOH aqueous solution and kept at 80 °C for 24 h to remove the silica template, then centrifuged, washed and dried at 80 °C. This sample was denoted as H-Sn/CeO2. H-CeO2 was also synthesised following the same route except the presence of SnCl4·5H2O and ammonia solution.
For comparison, the usual Sn/CeO2 catalyst was synthesized by co-precipitation method using Ce(NO3)3·6H2O and SnCl4·5H2O as precursors and 1 M (NH4)2CO3 as precipitator. This sample was denoted as SnO2–CeO2. Pure CeO2 was also prepared by the same method for parallel experiment purpose and denoted as CeO2.
2.2 Characterization
The powder X-ray diffraction (XRD) was performed with a Rigaku D/MAX-2200 X-ray diffractometer by using Cu Kα (40 kV, 40 mA) radiation and a secondary beam graphite monochromator. The morphologies were observed by a scanning electron microscope (SEM, JEOL JSM-6700F) and a field emission high resolution transmission electron microscope (HRTEM, JEOL JEM-2100F). Laser Raman spectroscopy (LRS) analysis was performed on a LabRAM Aramis (Japan Horiba) Laser Raman spectroscopy using Ar+ laser beam. The nitrogen adsorption–desorption isotherms of the samples were measured at 77 K using an ASAP 2020 volumetric adsorption analyzer. Before the measurements, all samples were degassed overnight at 473 K in a vacuum line. The specific surface area and the pore volume of the samples were calculated by the Brunauer–Emmett–Teller (BET) method, and the pore size distributions were derived from the desorption branches of isotherms using the Barrett–Joyner–Halenda (BJH) model. The X-ray photoelectron spectroscopy (XPS) was recorded on a Perkin-Elmer PHI 5000C ESCA system equipped with a dual X-ray source, using the Mg Kα (1253.6 eV) anode and a hemispherical energy analyzer. The back ground pressure during data acquisition was kept below 10−6 Pa. All binding energies were calibrated using contaminant carbon (C 1s = 284.6 eV) as a reference.Temperature-programmed reduction by hydrogen (H2-TPR) was obtained on a Tianjin XQ TP5080 auto-adsorption apparatus. 50 mg of the calcined catalyst was outgassed at 300 °C under N2 flow. After cooling to room temperature under N2 flow, the flowing gas was switched to 5% H2/N2, and the sample was heated to 980 °C at a ramping rate of 10 °C min−1. The H2 consumption was monitored by a TCD. Temperature-programmed desorption experiments of ammonia (NH3-TPD) were conducted on a Tianjin XQ TP5080 auto-adsorption apparatus. Before TPD, each sample was pretreated under He flow at 300 °C for 0.5 h, then saturated with high-purity anhydrous ammonia at 100 °C for 1 h and subsequently flushed at the same temperature for 1 h. Finally, the TPD operation was carried out from 100 to 800 °C at a heating rate of 10 °C min−1. The amount of NH3 desorbed was monitored by a TCD.
2.3 Catalytic activity measurements
The NH3-SCR activity measurement was carried out in a fixed-bed quartz micro-reactor operating in a steady state flow mode. The temperature of the reactor was monitored and controlled by thermocouples that were inserted into the centre of the catalyst bed. 0.4 g of catalysts were sieved with 40–60 mesh and used in each test. The reactant gases were fed to the reactor by an electronic mass flow controller. The typical reactant gas composition was as follows: 500 ppm NO, 500 ppm NH3, 3 vol% O2, 200 ppm SO2 (when used), 8 vol% H2O (when used), and balance N2. The total flow rate was 250 mL min−1 and thus a GHSV of 45![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 h−1 was obtained. The temperature increased from 125 to 400 °C. At each temperature step the data were recorded when the SCR reaction reached steady state after 15 min. The concentration of NO in the inlet and outlet gas was measured by a KM9106 flue gas analyzer. NO conversion was calculated according to the following expression:
000 h−1 was obtained. The temperature increased from 125 to 400 °C. At each temperature step the data were recorded when the SCR reaction reached steady state after 15 min. The concentration of NO in the inlet and outlet gas was measured by a KM9106 flue gas analyzer. NO conversion was calculated according to the following expression:
 | (1) |
3. Results and discussion
3.1 Characteristics
The TEM and SEM analysis was performed to investigate morphological characteristics of the H-Sn/CeO2 as shown in Fig. 2. Arrays of highly ordered mesoporous structure could be easily observed in Fig. 2a, which could provide great mass transfer ability and large surface area for the catalytic reaction. | ||
| Fig. 2 (a) and (b) TEM, (c) SEM and (d)–(f) EDX-mapping images of the H-Sn/CeO2. | ||
The TEM images in Fig. 2b further show that there are various pores in H-Sn/CeO2: the ∼4.0 nm pores, the 10–30 nm pores and the 70 nm pores (highlighted with red circle), indicating the existence of hierarchically macro-/meso-pores. It is well known that KIT-6 consists of two interpenetrating mesoporous channels linked by microporous channels.47 The branches of the two different sets of mesoporous channels in KIT-6 are separated by a thin silica wall.48 When both sets of the KIT-6 mesochannels are occupied by CeO2 and then the KIT-6 is dissolved away, the remaining pore is about 4 nm. When CeO2 grows in only one set of mesochannels and then the silica between them etched away, the remaining metal oxide has pores in the range of 10–30 nm. CeO2 will grow in only a proportion of one set of the KIT-6 mesochannels when the comprehensive destruction of the microchannels in KIT-6 by K+ occurs, leading to the formation of macropores (about 70 nm).49 The SEM image of the H-Sn/CeO2 (Fig. 2c) clearly exhibits that the ordered structure was formed by the accumulation of ∼20 nm-sized particles. The elemental mapping was performed to illustrate the spatial distribution of Ce and Sn species in the H-Sn/CeO2 (Fig. 2a). As revealed in Fig. 2d–f, the elemental mapping results confirm the uniform distribution of Ce and Sn species within the H-Sn/CeO2. The morphology and structure of the H-CeO2 are similar to those of H-Sn/CeO2, suggesting the universality of the preparation method (Fig. S1, ESI†).
The hierarchical nanostructure can also be further confirmed by N2 adsorption–desorption measurements, as shown in Fig. 3. As can be observed, the adsorption isotherms and hysteresis loops of the H-CeO2 and H-Sn/CeO2 seem to be of type IV and type H3, which is the characteristic feature of mesoporous solids, agreeing well with the TEM results.50 For CeO2 and SnO2–CeO2, the isotherms also show the mesoporous features, which could be resulted from the packing of nanoparticles. As illustrated in the inset of Fig. 3, the pore distribution of H-CeO2 and H-Sn/CeO2 display sharp peaks in the range of the mesopore and macropore, which prove the existence of hierarchical nanostructure. For the CeO2 and SnO2–CeO2, both of the pore distribution profiles show a sharp peak at 3–5 nm. By calculating the N2 adsorption–desorption isotherms, the specific surface area, pore size distribution and pore volume of the catalysts were obtained and summarized in Table 1. The BET surface area of the H-Sn/CeO2, H-CeO2, SnO2–CeO2 and CeO2 catalysts are calculated to be 157.4 m2 g−1, 207.8 m2 g−1, 98.2 m2 g−1 and 93.2 m2 g−1, respectively. Obviously, the BET surface area of the catalysts prepared by the template method is higher than that of the other two catalysts, which can be due to the contribution of hierarchical nanostructure. It is worth noting that the BET surface area of the H-Sn/CeO2 is smaller than that of the H-CeO2, which might be contributed to that the acidic SnCl4·5H2O precursor could destroy the ordered structure and led to a decrease in the surface area. The larger specific surface area and higher pore volume of these two hierarchical catalysts are beneficial to the adsorption, storage and diffusion of reactants, resulting in the enhancement of NH3-SCR activity.41
 | ||
| Fig. 3 N2 adsorption–desorption isotherms and pore size distributions (inset) of various catalysts. | ||
| Catalyst | BET surface area (m2 g−1) | Pore volume (cm3 g−1) | Pore size (nm) |
|---|---|---|---|
| H-Sn/CeO2 | 157.4 | 0.43 | 17.1 |
| H-CeO2 | 207.8 | 0.48 | 3.4 |
| SnO2–CeO2 | 98.2 | 0.12 | 19.1 |
| CeO2 | 93.2 | 0.18 | 3.8 |
The XRD experiments were performed to determine the crystalline nature of the samples. The XRD patterns of different catalysts are shown in Fig. 4a. All the catalysts show characteristic reflections at 2θ = 28.6°, 33.1°, 47.5°, 56.3°, 59.1°, 69.4°, 76.7°, 79.1° and 88.4° corresponded to the (111), (200), (220), (311), (222), (400), (331), (420) and (422) phase structure of CeO2 (JCPDS 34-0394), respectively. No evidence of Sn-containing phases was detected for the SnO2–CeO2 and H-Sn/CeO2 catalysts, suggesting that the Sn species were in an amorphous form or highly dispersed in the catalysts. The above results indicate that the primary crystalline phase of these four catalysts is CeO2. It is worth noting that a shift of the XRD peaks can be found for H-Sn/CeO2, which might be ascribed to the doping of Sn to the CeO2 lattice. Besides, weaker and broader diffraction peaks were observed, which could be attributed to the better dispersion of Ce and Sn species in H-Sn/CeO2. In addition, the EDS spectrum of the H-Sn/CeO2 is also performed in Fig. 4b, which reveals that the sample contains elements Ce, Sn and O, thus implies that the hierarchical nanostructure in Fig. 2a is a kind of Ce–Sn composite oxides.
 | ||
| Fig. 4 (a) XRD patterns of various catalysts and (b) EDX pattern of the H-Sn/CeO2. | ||
The Raman spectroscopy was carried out to obtain more information regarding the crystalline structure of the catalysts. Fig. 5 shows that the profiles for all the samples are dominated by the peak at around 465 cm−1, due to F2g mode of Ce–O vibration.51 As can be seen from the enlarged spectra, the peaks of SnO2–CeO2 and H-Sn/CeO2 locate at 467 cm−1 and 464 cm−1, respectively. Compared with the other two catalysts, the peaks of the Sn-doped catalysts shift slightly to low wave number direction and the width of the peaks increases, which can be attributed to that the Sn atoms incorporate into the crystal lattice of ceria. This is in accordance with the results of XRD and EDS. It has been demonstrated that the doping of hetero-atoms to ceria could lead to the generation of extra oxygen vacancies, which may enhance the redox ability of the catalyst and further facilitate NO oxidation to NO2, then improves the SCR activity via the “fast SCR” reaction.52,53
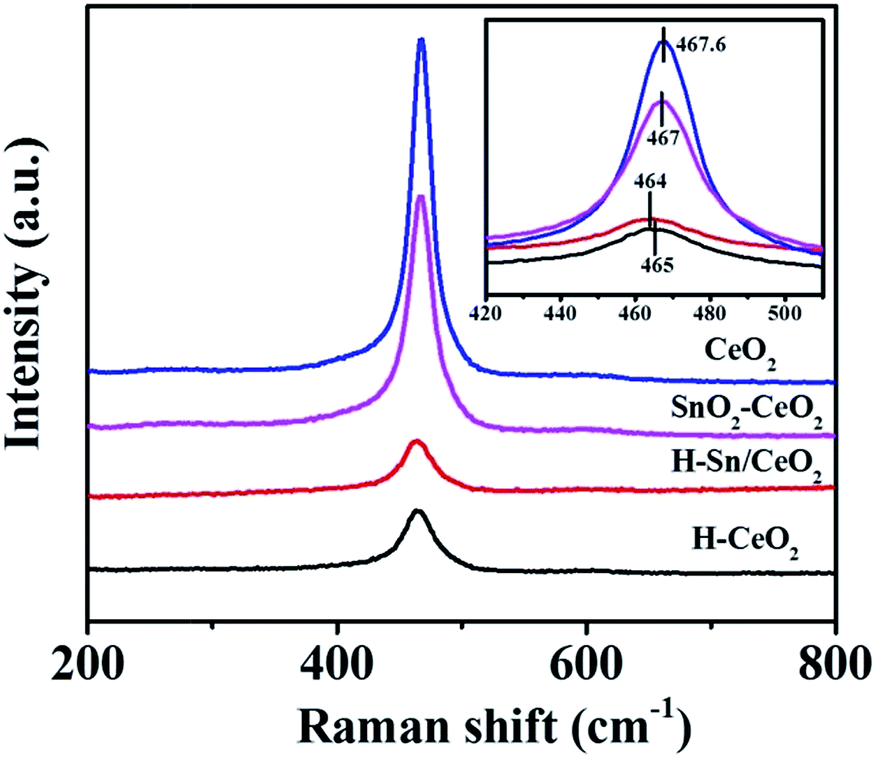 | ||
| Fig. 5 Raman spectra of various catalysts (inset: enlarged spectra). | ||
The XPS measurements were conducted to provide the information on the metal oxidation states and the content of the layers close to the surface of catalysts. Complicated XPS spectra of Ce 3d for different catalysts are presented in Fig. 6a. The surface atomic concentrations of Sn, Ce, O and the relative concentration ratios of different oxidation states are summarized in Table 2. By performing peak-fitting deconvolutions, the Ce 3d XPS spectra can be divided into ten characteristic peaks: u′′′ (916.6 eV), u′′ (907.5 eV), u′ (903.5 eV), u (901.1 eV), uo (899.0 eV), v′′′ (898.3 eV), v′′ (888.8 eV), v′ (884.9 eV), v (882.5 eV), vo (880.5 eV). The series of peaks labelled u and v represent the Ce 3d5/2 and Ce 3d3/2 states contribution, respectively.54 As reported previously, the uo, u′, vo and v′ peaks were assigned to Ce3+ chemical state while the u′′′, u′′, u, v′′′, v′′ and v peaks were attributed to Ce4+ chemical state.17 By calculating based on the area ratio of the Ce3+ species, the amounts of Ce3+ species relative to total Ce are observed to be 47.5% for H-Sn/CeO2, a bit less, at 45.6% for H-CeO2, and much less, at 43.4% for SnO2–CeO2 and at 42.3% for CeO2. The Ce3+/(Ce3+ + Ce4+) ratio of the hierarchical nanostructure catalysts are higher than those of other two catalysts, which means the relatively high Ce3+ content. This phenomenon could be explained by the hierarchical nanostructure and better dispersion of active species leading to more exposed Ce3+. Moreover, it can be seen that the doping of Sn also result in more Ce3+ species. It has been demonstrated the presence of Ce3+ is associated with the formation of oxygen vacancies according to charge balance, hence replenishing gaseous oxygen such as chemisorbed oxygen, which is very beneficial to the SCR process.55,56
 | ||
| Fig. 6 XPS spectra of (a) Ce 3d, (b) O 1s and (c) Sn 3d of various catalysts. | ||
| Catalyst | Sn (at%) | Ce (at%) | Ce3+/(Ce3+ + Ce4+) (%) | O (at%) | Oα/(Oα + Oβ) (%) |
|---|---|---|---|---|---|
| H-Sn/CeO2 | 0.96 | 6.39 | 47.5 | 43.77 | 55.2 |
| H-CeO2 | — | 6.87 | 45.6 | 44.32 | 38.2 |
| SnO2–CeO2 | 4.47 | 7.15 | 43.4 | 47.57 | 34.5 |
| CeO2 | — | 8.33 | 42.3 | 46.57 | 23.5 |
The O 1s XPS results of the catalysts are shown in Fig. 6b. The O 1s bands are deconvoluted by the curve-fitting procedure. The sub-bands at low binding energy (528.7–530.9 eV) assign to the lattice oxygen O2− (denoted as Oβ), and the sub-bands at high binding energy (531.4–532.5 eV) correspond to the chemisorbed oxygen (denoted as Oα), such as O− or O22− belonging to defect-oxide or hydroxyl-like group.57 As shown in Table 2, the Oα/(Oα + Oβ) ratio on H-Sn/CeO2 and H-CeO2 are 55.2%, 38.2%, respectively, higher than that on SnO2–CeO2 (34.5%) and CeO2 (23.5%). Moreover, the Sn-doped catalysts also create more Oα than the pure ceria catalysts. These results can be ascribed to the following: (1) the hierarchical nanostructure catalysts present larger specific surface area as well as better dispersion of active species and hence result in more exposed active oxygen species; and/or (2) there might be synergistic interaction of Sn and Ce species, leading to the formation of Ce3+, subsequently increase the amount of chemisorbed oxygen. Table 2 shows that the surface Oα and Ce3+ ratio of the catalysts follow the same trend. Previous reports have found that the chemisorbed oxygen species are more active than the lattice oxygen species, attributed to the higher mobility of Oα. Therefore, high relative concentration ratio of Oα/(Oα + Oβ) can promote the oxidation of NO to NO2 and subsequently facilitate the “fast SCR” reaction.27 It is conceivable that different Oα content on the catalysts will allow the distinction on the NH3-SCR performance.
Fig. 6c presents the Sn 3d XPS spectra of H-Sn/CeO2 and SnO2–CeO2. The Sn 3d XPS spectra consist of double peaks (Sn 3d5/2 486.5 eV and Sn 3d3/2 494.9 eV). The signal from Sn 3d5/2 shows an asymmetric peak, which indicates the coexist of Sn2+ and Sn4+.58 It has been reported that the synergetic interaction of Sn and Ce species via the redox equilibrium of 2Ce4+ + Sn2+ ↔ 2Ce3+ + Sn4+ is beneficial to the generation of Ce3+ over the catalyst.59 From Table 2, it is clear that the Ce3+ ratio on H-Sn/CeO2 is higher than that on the SnO2–CeO2, suggesting stronger synergetic effect in the former catalyst. Furthermore, the spectrum of H-Sn/CeO2 shift to a higher binding energy compared to SnO2–CeO2, indicating that part of the high valence Sn species existed over the H-Sn/CeO2 sample. This result is consistent with the Ce 3d XPS spectra. In addition, spectrum intensity of the H-Sn/CeO2 sample is weaker than that of the SnO2–CeO2, which means less surface Sn species on the H-Sn/CeO2, demonstrated by Table 2. This could be attributed to more Sn atoms incorporated into the CeO2 lattice and thus better dispersion of Sn species.
As is well known, the redox property of catalysts is remarkably related to the catalytic cycle in NH3-SCR of NO. The H2-TPR technique was employed to investigate the reducibility of the catalysts, and the obtained H2-TPR profiles are illustrated in Fig. 7. All of the H2-TPR profiles of the samples present three distinct H2 consumption peaks. In terms of CeO2, the reduction peaks at 447 °C, 524 °C and 866 °C can be observed while for H-CeO2 the reduction peaks locate at 420 °C, 578 °C and 766 °C, assigned to the reduction of surface, subsurface and bulk CeO2, respectively.29 Previous studies have demonstrated that the H2-TPR profile of pure SnO2 presents a strong reduction peak around 630 °C, ascribed to the reduction of Sn4+ to metal Sn.59,60 It is evident that the reduction peaks of the Sn-doped catalysts shift to much lower temperature compared to corresponding ceria catalysts, at 358 °C, 505 °C, 832 °C for SnO2–CeO2, and at 358 °C, 535 °C, 766 °C for H-Sn/CeO2. The reduction process of Sn species is overlapped with that of Ce species. The lower reduction peaks suggest the better redox ability of SnO2–CeO2 and H-Sn/CeO2. Thus, it is reasonable to deduce that there is a synergetic interaction between Ce and Sn species, which is in good agreement with the XPS analysis.36 It can be concluded that the doping of Sn element could improve the reducibility of the catalysts, leading to the enhancement of catalytic cycle in the NH3-SCR of NO. Besides, the TPR profiles show that the hierarchical nanostructure did not have direct impact on promoting the reducibility.
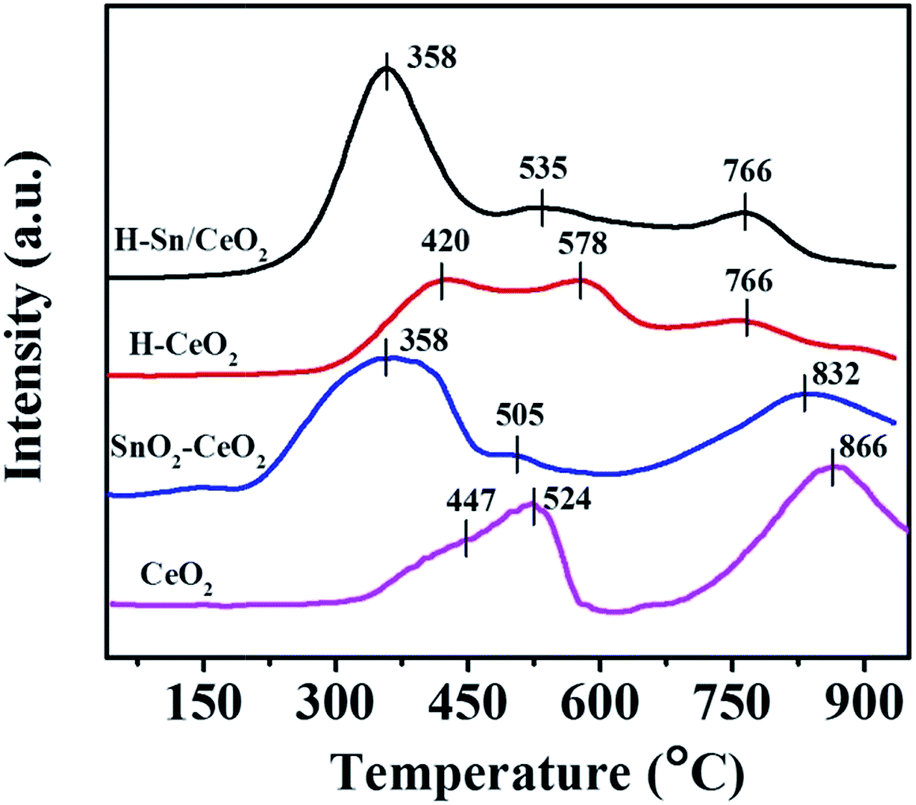 | ||
| Fig. 7 H2-TPR profiles of various catalysts. | ||
The NH3-TPD was conducted to investigate the surface acid amount and strength of the samples, and the obtained NH3-TPD profiles of different catalysts are shown in Fig. 8. The NH3-TPD profiles of H-Sn/CeO2 and H-CeO2 both show two desorption peaks: the NH3 desorption peak centered at 210 °C assigned to weak acid sites and that centered at 385 °C attributed to strong acid sites. Whereas, for SnO2–CeO2 and CeO2, one weak desorption peak at around 180 °C is observed, with a couple of extremely weak desorption peaks present in medium and high temperature region. It is well known that the position and area of desorption peak are relevant to the acid strength and acid amount, respectively.61 As compared with SnO2–CeO2 and CeO2, the desorption peaks of H-Sn/CeO2 and H-CeO2 obviously shift to the high-temperature region, which suggests that the strength of acid sites on H-Sn/CeO2 and H-CeO2 is stronger than that of SnO2–CeO2 and CeO2. The doping of Sn species into ceria also make contribution to the move of the desorption peaks toward to high temperature, indicating that the doped-Sn could enhance the acid strength of the ceria catalysts.52 On the other hand, the NH3-TPD profiles of H-Sn/CeO2 and H-CeO2 reveal much larger area, indicating the presence of abundant acid sites. Based on the TEM and N2 sorption results, the higher number of acid sites over H-Sn/CeO2 and H-CeO2 could be attributed to the larger specific surface area and characteristic of the hierarchical structures. In addition, the peak area of H-Sn/CeO2 and SnO2–CeO2 are slightly larger than that of corresponding ceria catalysts due to the acidic characteristic of Sn species. Therefore, the stronger strength and larger number of acid sites over the H-Sn/CeO2 will be benefit to the adsorption and activation of NH3 on the surface of the catalyst, and then give rise to the outstanding catalytic performance.
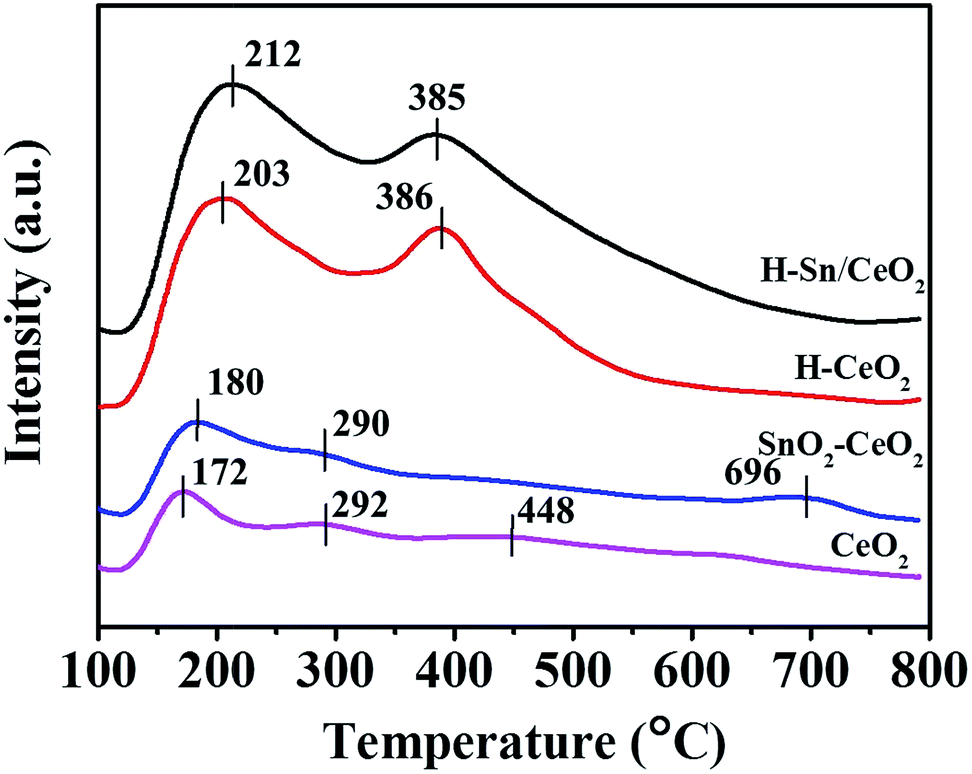 | ||
| Fig. 8 NH3-TPD profiles of various catalysts. | ||
3.2 Catalytic activity
The corresponding NH3-SCR performance of all catalysts, plotted as a function of temperature, is depicted in Fig. 9a. It is found that the NO conversion over all the catalysts increases first and then decreases with the temperature rising, as well as a significant difference on the catalytic activity of the catalysts is observed. The NO conversion of the CeO2 catalyst reaches its maximum value (about 70%) until 340 °C. As expected, the addition of Sn could effectively enhance the SCR activity as the SnO2–CeO2 catalyst shows superior performance than the CeO2.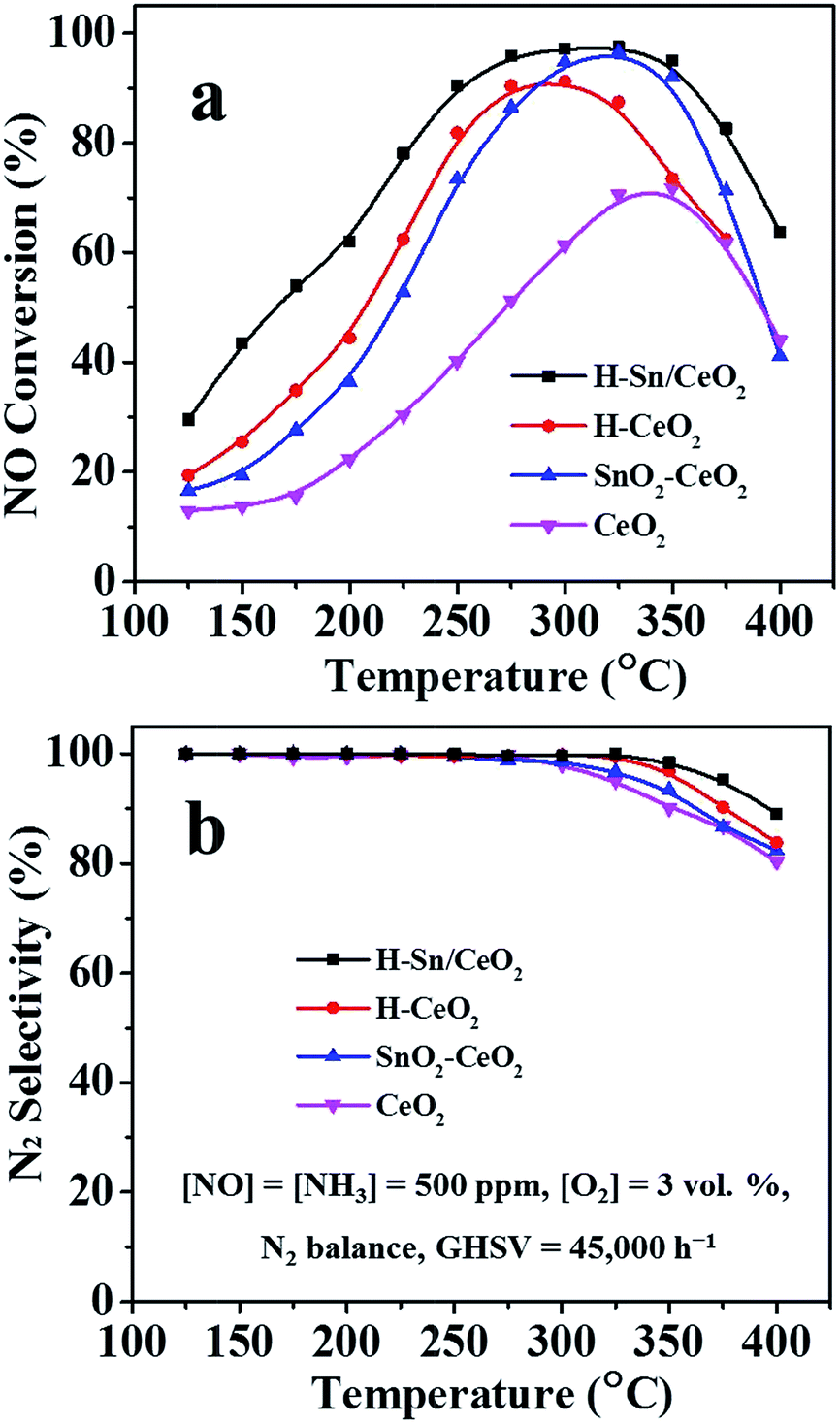 | ||
| Fig. 9 (a) NO conversion and (b) N2 selectivity of various catalysts. | ||
Specifically, the low and medium-temperature activity is greatly improved and a more extensive operating temperature window for NH3-SCR can be demonstrated. The SnO2–CeO2 catalyst gives the highest NO conversion (95%) at around 320 °C. Likewise, the H-CeO2 presents better catalytic performance in low and medium-temperature range compared to CeO2, obtaining 90% of NO conversion at 290 °C. Unlike the other catalysts, the H-Sn/CeO2 catalyst reveals higher SCR activity under the identical operating conditions. More than 90% of NO conversion is achieved in the temperature range of 250–360 °C, showing a rather broad temperature window for NH3-SCR process. Based on these results, it could be conjectured that firstly, the addition of Sn can not only enhance the low and medium-temperature activity of ceria catalyst but also improve its high temperature performance and secondly, the hierarchical nanostructure with better dispersion of active components has a significant beneficial effect on the NH3-SCR reactions.33,41 Besides, the N2 selectivity of H-Sn/CeO2 and H-CeO2 is higher than that of SnO2–CeO2 and CeO2 (Fig. 9b). The H-Sn/CeO2 exhibits the highest N2 selectivity in the full operating temperature range. Moreover, when Sn element is introduced, the enhancement in N2 selectivity for ceria catalysts can also observed, which agrees well with the previous reports.62
The diversity of catalytic activity over the catalysts could be attributed to the characteristic of hierarchical nanostructures with uniform active species dispersion as well as the interaction between Ce and Sn species. The TEM, SEM and N2 physisorption results of the H-Sn/CeO2 indicates the existence of hierarchically macro-/meso-pores with large specific surface area and good dispersion of elements, hence resulting in great mass transfer ability and more exposed active sites, which could be in favour of the excellent catalytic activity. The XRD and Raman analyses show that the Sn atoms incorporate into the crystal lattice of ceria, which might lead to synergistic interaction of Sn and Ce species. The XPS and H2-TPR results demonstrate the presence of synergistic interaction between uniformly dispersed Sn and Ce species in the H-Sn/CeO2, and thus result in more surface Ce3+ and Oα species and improved reducibility, which could promote the SCR reaction and enhance the SCR activity. The NH3-TPD results demonstrate that the H-Sn/CeO2 catalyst possesses more acid sites and stronger acid strength than the other three catalysts, which could facilitate the adsorption and activation of NH3. Based on these favorable properties, the H-Sn/CeO2 catalyst exhibits the excellent performance in the NH3-SCR reaction.
In practical application, the stability and H2O tolerance of catalysts are also important factors for evaluating the performance of catalysts in the NH3-SCR reaction. Fig. 10a illustrates the stability and H2O tolerance tests of the H-Sn/CeO2 as a function of time at a typical temperature 275 °C. The feed gas consists of 500 ppm NO, 500 ppm NH3, 8 vol% H2O (when used), 3 vol% O2, and balance gas N2 and the total flow rate is 250 mL min−1. During the stability test period, the NO conversion is maintained at about 96%. More importantly, the morphologies and structure of H-Sn/CeO2 are retained well after the stability test as shown in the TEM image (Fig. S2a, ESI†). Fig. S2b† depicts the N2 adsorption–desorption curves of the H-Sn/CeO2 after the stability test and the adsorption isotherms as well as hysteresis loops still show the characteristic feature of mesoporous solids. The corresponding BET surface area is calculated to be 157.6 m2 g−1, which is almost the same as that of the fresh catalyst. The pore size distribution of H-Sn/CeO2 is presented in the inset of Fig. S2b.† The pattern displays sharp peaks in a wide range of pore sizes, confirming the existence of mesopores and macropores. Thus, the observation evidently suggests that the H-Sn/CeO2 catalyst with hierarchical nanostructure is considerably stable to support the deNOx process.
 | ||
| Fig. 10 (a) Stability test and H2O resistance study (inset) of the H-Sn/CeO2 catalyst and (b) SO2 resistance study of the catalysts at 275 °C. | ||
The inset of Fig. 10a shows the H2O resistance test of the H-Sn/CeO2 catalyst. The NO conversion is 96% in the absence of H2O. The introduction of steam did not induce decline of the NO conversion over the catalyst, indicating that the H-Sn/CeO2 has a good capacity for water resistance. It has been reported that the inhibition of H2O on the catalyst surface is reversible, caused by the competitive adsorption between H2O and NH3 molecules on the active sites.63 The NH3-TPD analysis shows that there are abundant acid sites over the H-Sn/CeO2, which might preferentially absorb NH3 other than H2O from the gas phase and then be responsible for its excellent H2O resistance.
It is well known that even though passing through the desulfurization device, the exhaust fumes still contain trace amounts of SO2 (about 100 ppm), which can induce the catalyst poisoning and deactivation. Therefore, the impact of SO2 on the NH3-SCR activity of the H-Sn/CeO2 and H-CeO2 was also investigated. Fig. 10b depicts the catalytic performance of these two catalysts, as a function of time in the presence of 200 ppm SO2 at a typical temperature of 275 °C. In the absence of SO2, the NO conversion over the H-Sn/CeO2 and H-CeO2 is 96% and 90%, respectively. After introducing 200 ppm SO2, the NO conversion both experience a slight increase in the first few hours and then decrease gradually to a steady state. In the case of H-Sn/CeO2, the NO conversion slowly drops to 78% and remains stable. Upon switching off the SO2 from the feed gas, the NO conversion is gradually restored to 81%, which has declined by 15% compared to the initial value. Meanwhile, the NO conversion over the H-CeO2 keeps lowering down and stand stable at 60%. After stopping the SO2 supply, the NO conversion can be partly recovered and finally maintained at 64%, which is 26% less than the initial conversion, indicating more severe poisoning by SO2 for H-CeO2. These results show that the doping of Sn element can effectively improve the SO2-resistance of ceria catalysts, which is in good conformity to the previous reports.36 The transient increase of NO conversion after introducing SO2 might be because SO2 reacts with NH3 over catalyst surface and the generated surface sulfates can work as new acid sites to promote the NH3-SCR reactions.64 With constant introduction of SO2, more and more ammonium sulfate species are generated and deposited on the catalyst surface, blocking the active sites of the catalyst surface, leading to the gradually decrease of catalytic activity. The final NO conversion of these two catalysts could not be restored to their initial values, which could be ascribed to the irreversible deactivation caused by the stable metal sulfates and/or NH4HSO4.28,65
4. Conclusions
The hierarchically macro-/mesoporous Sn/CeO2 catalyst was successfully created by using KIT-6 as hard template for selective catalytic reduction of NO with NH3. In comparison with H-CeO2, SnO2–CeO2 and CeO2, the H-Sn/CeO2 catalyst with uniform element composition and dispersion shows greatly improved NH3-SCR activity as well as more extensive operating temperature window. The NO conversion of H-Sn/CeO2 is higher than 90% in the range of 250–360 °C with better N2 selectivity. 96% of NO conversion can be maintained during the stability test period and the hierarchical nanostructure was retained well. The H-Sn/CeO2 catalyst also exhibits excellent water resistance and enhanced SO2 tolerance. The outstanding catalytic performance of the H-Sn/CeO2 can be ascribed to the following points. On the one hand, the hierarchical nanostructure catalysts possess abundant mesopores and macropores as well as large specific surface area, hence resulting in more exposed active species and acid sites, which is conducive to the adsorption, storage and diffusion of reactants. On the other hand, the synergistic interaction of Sn and Ce species due to the incorporation of Sn to the ceria lattice gives rise to more surface Ce3+ and Oα species along with improved reducibility. The doping of acidic Sn leads to stronger strength and larger number of acid sites over the H-Sn/CeO2 as well. In summary, the H-Sn/CeO2 catalyst should be a promising candidate for the elimination of NO, and the hierarchical nanostructure materials can be applied broadly in many fields of environmental protection.Acknowledgements
The authors acknowledge the support of the National Basic Research Program of China (973 Program, 2014CB660803) and the Shanghai Municipal Education Commission (14ZZ097). We thank Mr Y. L. Chu from the Analysis and Test Center of SHU for help with the TEM and SEM measurements.References
- N. Y. Topsøe, Science, 1994, 265, 1217–1219 Search PubMed.
- C. H. Kim, G. S. Qi, K. Dahlberg and W. Li, Science, 2010, 327, 1624–1627 CrossRef CAS PubMed.
- P. G. Smirniotis, D. A. Peña and B. S. Uphade, Angew. Chem., 2001, 40, 2479–2482 CrossRef CAS.
- A. T. Krishnan and A. L. Boehman, Appl. Catal., B, 1998, 18, 189–198 CrossRef CAS.
- P. G. W. A. Kompio, A. Brückner, F. Hipler, G. Auer, E. Löffler and W. Grünert, J. Catal., 2012, 286, 237–247 CrossRef CAS.
- P. Forzatti, I. Nova and E. Tronconi, Angew. Chem., 2009, 121, 8516–8518 CrossRef.
- W. C. Wang, G. McCool, N. Kapur, G. Yuan, B. Shan, M. Nguyen, U. M. Graham, B. H. Davis, G. Jacobs, K. Cho and X. Hao, Science, 2012, 337, 832–835 CrossRef CAS PubMed.
- R. H. Gao, D. S. Zhang, X. G. Liu, L. Y. Shi, P. Maitarad, H. R. Li, J. P. Zhang and W. G. Cao, Catal. Sci. Technol., 2013, 3, 191–199 CAS.
- S. B. Kristensen, A. J. Kunov-Kruse, A. Riisager, S. B. Rasmussen and R. Fehrmann, J. Catal., 2011, 284, 60–67 CrossRef CAS.
- L. Huang, X. Zhao, L. Zhang, L. Shi, J. Zhang and D. Zhang, Nanoscale, 2015, 7, 2743–2749 RSC.
- S. Djerad, M. Crocoll, S. Kureti, L. Tifouti and W. Weisweiler, Catal. Today, 2006, 113, 208–214 CrossRef CAS.
- J. P. Dunn, P. R. Koppula, H. G. Stenger and I. E. Wachs, Appl. Catal., B, 1998, 19, 103–117 CrossRef CAS.
- J. Han, D. S. Zhang, P. Maitarad, L. Y. Shi, S. X. Cai, H. R. Li, L. Huang and J. P. Zhang, Catal. Sci. Technol., 2015, 5, 438–446 CAS.
- L. Zhang, L. Y. Shi, L. Huang, J. P. Zhang, R. H. Gao and D. S. Zhang, ACS Catal., 2014, 4, 1753–1763 CrossRef CAS.
- H. Hu, S. X. Cai, H. R. Li, L. Huang, L. Y. Shi and D. S. Zhang, ACS Catal., 2015, 5, 6069–6077 CrossRef CAS.
- C. Fang, D. S. Zhang, L. Y. Shi, R. H. Gao, H. R. Li, L. P. Ye and J. P. Zhang, Catal. Sci. Technol., 2013, 3, 803–811 CAS.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 5162–5167 CrossRef CAS.
- D. S. Zhang, X. J. Du, L. Y. Shi and R. H. Gao, Dalton Trans., 2012, 41, 14455–14475 RSC.
- N. A. S. Amin, E. F. Tan and Z. A. Manan, Appl. Catal., B, 2003, 43, 57–69 CrossRef CAS.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, Chem. Commun., 2011, 47, 8046–8048 RSC.
- W. P. Shan, F. D. Liu, H. He, X. Y. Shi and C. B. Zhang, ChemCatChem, 2011, 3, 1286–1289 CrossRef CAS.
- G. S. Qi and R. T. Yang, Chem. Commun., 2003, 848–849 RSC.
- Y. Li, H. Cheng, D. Y. Li, Y. S. Qin, Y. M. Xie and S. D. Wang, Chem. Commun., 2008, 1470–1472 RSC.
- W. Q. Xu, Y. B. Yu, C. B. Zhang and H. He, Catal. Commun., 2008, 9, 1453–1457 CrossRef CAS.
- X. Gao, Y. Jiang, Y. C. Fu, Y. Zhong, Z. Y. Luo and K. F. Cen, Catal. Commun., 2010, 11, 465–469 CrossRef CAS.
- Y. S. Shen, S. M. Zhu, T. Qiu and S. B. Shen, Catal. Commun., 2009, 11, 20–23 CrossRef CAS.
- Z. B. Wu, R. B. Jin, Y. Liu and H. Q. Wang, Catal. Commun., 2008, 9, 2217–2220 CrossRef CAS.
- H. H. Phil, M. P. Reddy, P. A. Kumar, L. K. Ju and J. S. Hyo, Appl. Catal., B, 2008, 78, 301–308 CrossRef CAS.
- L. Zhang, W. X. Zou, K. L. Ma, Y. Cao, Y. Xiong, S. G. Wu, C. J. Tang, F. Gao and L. Dong, J. Phys. Chem. C, 2015, 119, 1155–1163 CAS.
- R. B. Jin, Y. Liu, Y. Wang, W. L. Cen, Z. B. Wu, H. Q. Wang and X. L. Weng, Appl. Catal., B, 2014, 148–149, 582–588 CrossRef CAS.
- Y. L. Wang, X. C. Jiang and Y. N. Xia, J. Am. Chem. Soc., 2003, 125, 16176–16177 CrossRef CAS PubMed.
- M. Epifani, J. D. Prades, E. Comini, E. Pellicer, M. Avella, P. Siciliano, G. Faglia, A. Cirera, R. Scotti, F. Morazzoni and J. R. Morante, J. Phys. Chem. C, 2008, 112, 19540–19546 CAS.
- X. F. Tang, J. H. Li, L. S. Wei and J. M. Hao, Chin. J. Catal., 2008, 29, 531–536 CrossRef CAS.
- Z. M. Liu, J. H. Li and J. M. Hao, Chem. Eng. J., 2010, 165, 420–425 CrossRef CAS.
- P. W. Park, H. H. Kung, D. W. Kim and M. C. Kung, J. Catal., 1999, 184, 440–454 CrossRef CAS.
- H. Z. Chang, J. H. Li, X. Y. Chen, L. Ma, S. J. Yang, J. W. Schwank and J. M. Hao, Catal. Commun., 2012, 27, 54–57 CrossRef CAS.
- X. L. Li, Y. H. Li, S. S. Deng and T. A. Rong, Catal. Commun., 2013, 40, 47–50 CrossRef.
- D. S. Zhang, L. Zhang, L. Y. Shi, C. Fang, H. R. Li, R. H. Gao, L. Huang and J. P. Zhang, Nanoscale, 2013, 5, 1127–1136 RSC.
- C. Y. Ma, Z. Mu, J. J. Li, Y. G. Jin, J. Cheng, G. Q. Lu, Z. P. Hao and S. Z. Qiao, J. Am. Chem. Soc., 2010, 132, 2608–2613 CrossRef CAS PubMed.
- G. Li and R. C. Jin, Acc. Chem. Res., 2013, 46, 1749–1758 CrossRef CAS PubMed.
- Y. N. Shi, S. Chen, H. Sun, Y. Shu and X. Quan, Catal. Commun., 2013, 42, 10–13 CrossRef CAS.
- F. Jiao, A. H. Hill, A. Harrison, A. Berko, A. V. Chadwick and P. G. Bruce, J. Am. Chem. Soc., 2008, 130, 5262–5266 CrossRef CAS PubMed.
- Y. H. Zhang, X. C. Zhao, Y. Wang, L. K. Zhou, J. Y. Zhang, J. Wang, A. Q. Wang and T. Zhang, J. Mater. Chem. A, 2013, 1, 3724–3732 CAS.
- J. H. Huang, Z. Q. Tong, Y. Huang and J. F. Zhang, Appl. Catal., B, 2008, 78, 309–314 CrossRef CAS.
- J. Yu, F. Guo, Y. L. Wang, J. H. Zhu, Y. Y. Liu, F. B. Su, S. Q. Gao and G. W. Xu, Appl. Catal., B, 2010, 95, 160–168 CrossRef CAS.
- W. Li and D. Y. Zhao, Chem. Commun., 2013, 49, 943–946 RSC.
- F. Kleitz, S. Hei Choi and R. Ryoo, Chem. Commun., 2003, 2136–2137 RSC.
- T.-W. Kim, F. Kleitz, B. Paul and R. Ryoo, J. Am. Chem. Soc., 2005, 127, 7601–7610 CrossRef CAS PubMed.
- Y. Ren, Z. Ma, R. E. Morris, Z. Liu, F. Jiao, S. Dai and P. G. Bruce, Nat. Commun., 2013, 4, 2015 Search PubMed.
- P. Kumar, N. Mal, Y. Oumi, K. Yamana and T. Sano, J. Mater. Chem., 2001, 11, 3285–3290 RSC.
- B. M. Reddy, A. Khan, Y. Yamada, T. Kobayashi, S. Loridant and J. C. Volta, J. Phys. Chem. B, 2003, 107, 11475–11484 CrossRef CAS.
- H. Z. Chang, X. Y. Chen, J. H. Li, L. Ma, C. Z. Wang, C. X. Liu, J. W. Schwank and J. M. Hao, Environ. Sci. Technol., 2013, 47, 5294–5301 CrossRef CAS PubMed.
- Z. Y. Pu, J. Q. Lu, M. F. Luo and Y. L. Xie, J. Phys. Chem. C, 2007, 111, 18695–18702 CAS.
- F. Larachi, J. Pierre, A. Adnot and A. Bernis, Appl. Surf. Sci., 2002, 195, 236–250 CrossRef CAS.
- Y. Liu, W. Y. Yao, X. L. Cao, X. L. Weng, Y. Wang, H. Q. Wang and Z. B. Wu, Appl. Catal., B, 2014, 160–161, 684–691 CrossRef CAS.
- X. Wang, W. Wen, Y. Su and R. Wang, RSC Adv., 2015, 5, 63135–63141 RSC.
- F. D. Liu, H. He, Y. Ding and C. B. Zhang, Appl. Catal., B, 2009, 93, 194–204 CrossRef CAS.
- G. L. Xiao, S. A. Li, H. Li and L. Q. Chen, Microporous Mesoporous Mater., 2009, 120, 426–431 CrossRef CAS.
- L. Zhang, L. L. Li, Y. Cao, Y. Xiong, S. G. Wu, J. F. Sun, C. J. Tang, F. Gao and L. Dong, Catal. Sci. Technol., 2015, 5, 2188–2196 CAS.
- X. J. Yao, C. J. Tang, Z. Y. Ji, Y. Dai, Y. Cao, F. Gao, L. Dong and Y. Chen, Catal. Sci. Technol., 2013, 3, 688–698 CAS.
- L. Zhang, D. S. Zhang, J. P. Zhang, S. X. Cai, C. Fang, L. Huang, H. R. Li, R. H. Gao and L. Y. Shi, Nanoscale, 2013, 5, 9821–9829 RSC.
- M. Qiu, S. Zhan, D. Zhu, H. Yu and Q. Shi, Catal. Today, 2015, 258, 103–111 CrossRef CAS.
- P. P. Hu, Z. W. Huang, W. M. Hua, X. Gu and X. F. Tang, Appl. Catal., A, 2012, 437–438, 139–148 CAS.
- L. Zhang, L. L. Li, Y. Cao, X. J. Yao, C. Y. Ge, F. Gao, Y. Deng, C. J. Tang and L. Dong, Appl. Catal., B, 2015, 165, 589–598 CrossRef CAS.
- N. Tang, Y. Liu, H. Q. Wang and Z. B. Wu, J. Phys. Chem. C, 2011, 115, 8214–8220 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: TEM images, SEM image, N2 adsorption–desorption isotherms and pore size distribution of samples. See DOI: 10.1039/c6ra18339e |
| This journal is © The Royal Society of Chemistry 2016 |