
Structural evolution of palladium nanoparticles and their electrocatalytic activity toward ethanol oxidation in alkaline solution†
Changna Wena,
Zhaopeng Lia,
Caiyun Caoa,
Yiqian Wang*b,
Peizhi Guo*a and
X. S. Zhaoa
aInstitute of Materials for Energy and Environment, State Key Laboratory Breeding Based of New Fiber Materials and Modern Textile, School of Materials Science and Engineering, Qingdao University, Qingdao, 266071, P. R. China. E-mail: pzguo@qdu.edu.cn; qduguo@163.comn; Tel: +86 532 859 53982
bSchool of Physics, Qingdao University, Qingdao, 266071, P. R. China. E-mail: yqwang@qdu.edu.cn
First published on 12th September 2016
Abstract
A series of palladium nanoparticles with sizes ranging from 3 to 10 nm were synthesized with ethylene glycol as the solvent and reducing agent. Based on X-ray diffraction, transmission electron microscopy and high-resolution transmission electron microscopy characterization, the as-prepared palladium nanoparticles were well-crystalline and uniformly dispersed. Electrochemical measurements showed that the smallest (∼3 nm) palladium nanoparticles exhibited the highest current density and the best cycling stability and durability among the four kinds of palladium nanoparticles for the electrocatalytic oxidation of ethanol. In addition, small palladium nanoparticles were spherical while large nanoparticles with a certain decline in electrocatalytic activity showed distinct shape. The formation and shape evolution of the palladium nanoparticles as well as their structure–property relationship have been studied and analyzed based on the experimental data.
Introduction
In the past few years, noble-metal nanostructures have been of great interest, especially for their promising applications in fuel cells as efficient electrocatalysts, because of their high chemical activity and surface energy. Palladium (Pd) nanostructures in particular are considered to be the best alternative to Pt and have been extensively studied due to their relatively low cost, excellent electrocatalytic performance and strong anti-toxicity.1–3 The morphology, particle size and structure of the nanoparticle play vital roles in determining the catalytic activity of palladium nanoparticles (NPs).4–7 Hence, the preparation of palladium with well-controlled size and shape is the key for its application. At present, the preparation methods of nanomaterials are generally divided into physical processes (e.g., the gas-phase method, thermal-evaporation method and sputtering method) and chemical processes like the electrochemical method,8 emulsion method,9 hydrothermal method10 and sol–gel ethod.11–13 In general, physical processes are comparatively simple, but the particles are relatively large in size. As shown in previous reports, the morphologies of the Pd nanostructures obtained through chemical processes are nanocubes,14 nanosheets,15 nanotubes,16,17 etc. Nevertheless, the synthesis and properties of highly active Pd NPs with certain sizes and shapes as well as their widespread commercialization still face huge challenges for the moment.Among the synthetic strategies, the polyol process, as a convenient, effective and low-cost route, has drawn increasing attention to synthesize Pd NPs.18–21 In addition, polyvinylpyrrolidone (PVP) has a prominent effect on the morphology of metal nanoparticles due to the selective adsorption on different crystal surfaces that result in the anisotropic growth of metal nanostructures.22–24 Usually, there are four main processes of crystal growth: reduction, nucleation, growth and production. It is generally believed that the crystal shape can be controlled by manipulating the equilibrium relationship between the crystal nucleation and growth rates. The growth process of the crystal product is strongly dependent on the presence of an initial seed. If the initial crystal seed is a single crystal, the obtained products are mostly single crystals. However, if the initial crystal seed has a pointed octahedral shape, which has eight (111) crystal faces and six (100) crystal faces, the final products are mostly nanocubes.16 Many efforts have been put forth to take advantage of the polyol process.25 As demonstrated in previous studies, Ag nanowires have been synthesized by the reduction of AgNO3 with ethylene glycol using Pt or Ag as the crystal seed and PVP as the surfactant.26 Subsequently, Ag nanowire structures can also be synthesized in the absence of crystal seeds.27 Metal nanowires contact the PVP layer through the Ag–O layer, which also demonstrates the existence of multiple twinned crystals.28
The relationship between the structure and properties of Pd NPs still needs further exploration. In this paper, four types of Pd NPs with different morphologies and sizes were synthesized through a simple and effective method using ethylene glycol as the solvent and reduction agent. All the Pd NPs are well-dispersed with good crystalline nature. It is found that large Pd NPs show distinct shapes while small ones are typical of spheres. The formation mechanism of the Pd NPs is discussed based on structural characterization. The experimental data show that small-sized Pd NPs display better electrocatalytic activity towards the electrooxidation of ethanol than large ones.
Experimental
Materials and regents
All the chemicals used in this study, including palladium(II) chloride, sodium chloride, sulfuric acid, methanol, ethanol, ethylene glycol (EG), acetone and PVP (molecular weight of 58![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000), were of analytical grade and purchased from Sinopharm Chemical Reagent Company. Distilled water was deionized to a resistivity of 18.2 MΩ cm for electrochemical measurements. All of the reagents were used as received without further purification.
000), were of analytical grade and purchased from Sinopharm Chemical Reagent Company. Distilled water was deionized to a resistivity of 18.2 MΩ cm for electrochemical measurements. All of the reagents were used as received without further purification.
Synthesis of Pd NPs
Characterization
X-ray diffraction (XRD) experiments were carried out on a Bruker D8 Advance X-ray diffractometer equipped with CuKα radiation source (λ = 0.15418 nm) from 10° to 80° (2θ). Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) images were recorded on a JEM-2000EX (JEOL) and JEM-2100FEG (JEOL) transmission electron microscope operated at accelerating voltages of 160 and 200 kV, respectively.Electrochemical measurements
All the electrochemical measurements were conducted on a CHI760D workstation at ambient temperature with a three-electrode system. A platinum foil was used as the counter electrode, and a saturated calomel electrode (SCE) for acidic solutions or a mercury/mercuric oxide (Hg/HgO) electrode for alkaline solutions was used as the reference electrode. The working electrode was prepared using a glassy carbon electrode (GCE, 3 mm in diameter) as the substrate. Each Pd electrocatalyst was dispersed in ethanol with a concentration of 1 mg mL−1 under stirring. A volume of 10 μL catalyst ink was then drop-casted onto a glass carbon disk and dried in air to obtain a catalyst film as a typical electrode. The as-made electrode was activated in 1 mol L−1 H2SO4 solution at a scan rate of 100 mV s−1 and then operated in an electrolyte solution composed of 1 mol L−1 ethanol and 1 mol L−1 KOH for the electrocatalytic oxidation of ethanol at 50 mV s−1.Results and discussion
XRD analysis
Fig. 1 shows the XRD patterns of the four types of as-synthesized Pd NPs. As illustrated, three diffraction peaks of the Pd NPs were observed at 2θ values of 40.3°, 46.7° and 68.3°, corresponding to the (111), (200) and (220) crystal planes of Pd0, respectively, which identifies the formation of face-centered cubic (fcc) Pd NPs with high crystallinity (JCPDS no. 46-1043). It is noteworthy that all the diffraction peaks of the four kinds of Pd NPs were broad, indicating that the crystalline dimension of the as-prepared Pd NPs was rather small, and the average crystal sizes of Pd-a, Pd-b, Pd-c and Pd-d were calculated to be about 3.1, 5.2, 7.3 and 10.4 nm, respectively, based on the measurements of the strongest (111) diffraction peak in Fig. 1 according to the Scherrer formula. The larger Pd NPs displayed stronger diffraction intensities than the smaller ones. | ||
| Fig. 1 XRD patterns of Pd NPs: Pd-a (a), Pd-b (b), Pd-c (c) and Pd-d (d). | ||
Morphology and structure
Fig. 2 shows the representative TEM images of the four types of Pd NPs. It is obvious that all the Pd NPs prepared via direct reduction by the solvent ethylene glycol were well-defined and monodispersed, and almost no aggregation was observed. However, the dimensions of four types of Pd NPs were significantly different. More specifically, the particle size of the Pd-a sample was the smallest, about 3.4 ± 0.6 nm (Fig. 2A and B), based on the statistical result of the particle size distribution, which was collected from 250 NPs (Fig. 2B). Fig. 2C and D show that the particle size of Pd-b was about 5.0 ± 0.6 nm, slightly larger than that of Pd-a, indicating that the Pd NP size was slightly decreased when the synthetic time was increased from 20 min to 4 h. The size of the Pd-c NPs, which were obtained through secondary growth using Pd-a as the seed, was about 6.5 ± 0.5 nm (Fig. 2E and F), obviously larger than the sizes of Pd-a or Pd-b. Meanwhile, Pd-c NPs were almost in the shape of an octahedron. As a fcc metal, Pd nanocrystals can take a variety of geometrical shapes.23,29 The further growth of seeds might selectively enlarge one set of crystallographic facets at the expense of others to yield the final shape. Single-crystal seeds can evolve into cubes given a certain ratio of growth rates along the (111) and (110) directions.29 The obtained Pd-c NPs were then used as the seeds for the formation of Pd-d by adding them into the initial synthetic system that was used to prepare Pd-a NPs. The size of Pd-d was about 10 ± 1.0 nm, as derived from Fig. 2G and H. As the seed grew into a nanocrystal, the growth rates of the different facets could be altered by capping agents to control the final shape. If the initial crystal seed had a pointed octahedral shape with eight (111) crystal faces and six (100) crystal faces, the final products were mostly nanocubes.16 Compared with the former three Pd samples, the Pd-d NPs showed distinct shapes that were mainly cube structures accompanied with a small amount of triangles and polyhedral structures. The size of Pd-d was obviously larger than that of any of the former samples (Fig. 2H). All the above results reveal that Pd NPs can be synthesized in a controlled manner by manipulating the reaction time or the crystal seeds. Moreover, the TEM results for the four kinds of Pd NPs were in agreement with the XRD measurements. | ||
| Fig. 2 TEM images of Pd-a (A), Pd-b (C), Pd-c (E) and Pd-d (G) and particle size distribution plots of Pd-a (B), Pd-b (D), Pd-c (F) and Pd-d (H). The inset in each TEM image is the SAED pattern of the corresponding sample. | ||
The selected-area electron-diffraction (SAED) patterns of four Pd NPs, as shown in the insets of Fig. 2, are used to analyze their crystalline features. On the one hand, the emergence of the bright diffraction rings or spots in the SAED patterns confirms that all the Pd NPs were highly crystalline. On the other hand, the SAED pattern of Pd-a was an obvious diffraction ring (inset of Fig. 2A), because many Pd-a NPs could be detected and bombarded by electron beam in the electron diffraction test. Likewise, although we also observe annular diffraction patterns in the SAED pattern of Pd-d, most of which were composed of single bright diffraction spots, indicating that the Pd-d NP was single-crystalline (the inset of Fig. 2G).
Fig. 3 shows the HRTEM images of Pd-a, Pd-b, Pd-c and Pd-d. As illustrated in Fig. 3A and C, the as-prepared Pd-a and Pd-b were spherical particles. In contrast, the Pd-c (Fig. 3E) and Pd-d (Fig. 3G) samples presented clear polygonal or polyhedral structures after secondary and third growth, respectively, which matched the well-defined lattice fringes that are observed in Fig. 3G and H. That is, in spite of the presence of the protective agent PVP, the growth of large Pd NPs through the seed strategy might primarily depend on the crystal surface energy of small Pd NPs, for which the crystal grain grew quickly along the perpendicular direction relative to the crystal plane with high energy, thus leading the production of larger Pd-c and Pd-d NPs with regular shapes. Unfortunately, the morphologies of Pd-c and Pd-d were not uniform under the present experimental conditions. Based on the HRTEM images of the four kinds of Pd NPs (Fig. 3B, D, F and H), the lattice spacings of the samples were measured to be 2.35 to 2.40 Å and 2.53 to 2.56 Å, ascribed to the (111) and (200) planes of Pd, respectively, similar to the standard values of bulk Pd samples.30 In addition, the angle between the two crystal surfaces was measured to be about 53°, in accordance with the theoretical calculations.31 It can be considered that the (110) crystal plane of the as-synthesized Pd NPs was the axial plane, and the crystal structure had the characteristics of a single crystal.
 | ||
| Fig. 3 HRTEM images of Pd-a (A and B), Pd-b (C and D), Pd-c (E and F) and Pd-d (G and H). | ||
Formation mechanism of Pd NPs
The formation mechanisms of the Pd NPs were further studied by two types of experiments. On one hand, the effects of different reaction time on morphology were tested; on the other hand, the effect of the synthetic procedure was studied by the seed growth method.The intermediate products from the synthetic systems for Pd-a and Pd-b samples were collected from the initial reaction system after reaction times of 4 h and 20 min, respectively. The samples collected after the reaction times of 30 min (Pd-e), 60 min (Pd-f) and 120 min (Pd-g) were characterized by TEM measurement (Fig. S1†). The experimental results show that the reaction time had a slightly effect on the size of the Pd NPs, as shown in Fig. 2A, C and S1.† The dimensions of the nanocrystals gradually increased in the initial reaction stage and began to decrease with the sequential extension of reaction time. That is, the nanocrystals tended to grow when the reaction time was short on account of the high surface energies of small particles and large concentration of palladium precursor (Fig. 2A, S1A and B†). In addition, the existence of the polymer PVP in the synthetic system inhibited the aggregation of as-synthesized Pd nanoparticles.32 However, the slight decrease in particle size with extended reaction time might be attributed to the high reactivity of Pd NPs, which led a portion of them to participate in reaction, eventually forming nanoparticles with uniform size.
Additional experimental data were obtained by adjusting the synthetic procedures. As depicted in Fig. S2A,† Pd NPs with the sizes of about 5 to 8 nm (similar to Pd-c) were obtained when Pd-c nanoparticles in solution were used directly as the seeds. However, the Pd NPs were irregular and had a large size distribution compared to Pd-c. In contrast, if the collected solid Pd-b was used as the seed, as shown in Fig. S2B,† the particle size of the final product was larger than that of Pd-b. However, the size distribution was obviously uneven, likely due to the inconformity of the Pd-b surface during the re-dissolution process. Based on the above experimental results, Scheme 1 illustrates the schematic formation processes of the four types of Pd NPs. It is suggested that the sizes and shapes of the Pd NPs are determined by the surface energy and adsorption properties of the Pd nanocrystals, leading to the formation of large Pd NPs with regular shapes.33
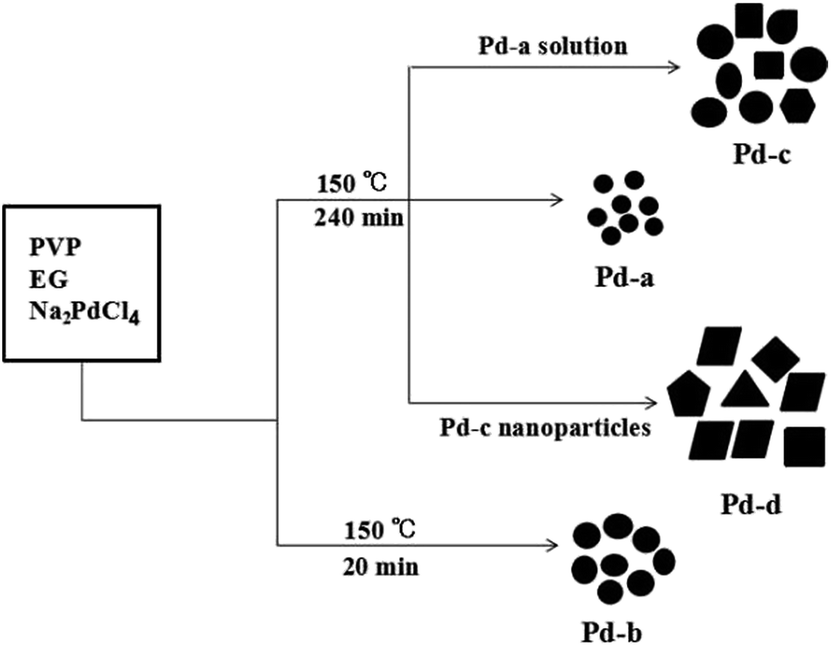 | ||
| Scheme 1 Schematic illustration of the formation mechanisms of Pd NPs based on the solution-based reduction method. | ||
Electrochemical characterization
Generally, Pd NPs exhibit excellent electrocatalytic activity toward the oxidation of alcohols. As shown in Fig. 4, the redox peaks displayed on the CV curves were in agreement with the characteristic peaks of Pd-modified electrodes reported in the literature.15,34–36 The sharp peak observed approximately at 0.43 V was attributed to the reduction peak of produced Pd oxide.37 In addition, the two peaks appearing at 0 to −0.12 V in the positive sweep direction were ascribed to the desorption and adsorption of H on the surfaces of the Pd NPs,38 which are usually used to estimate the electrochemically active surface areas (ECSAs). The ECSAs of the Pd-a, Pd-b, Pd-c and Pd-d NPs were calculated to be 15.0, 13.4, 9.80 and 6.96 cm2 mg−1, respectively, by the formula Q (μC)/424 cm2, in which Q stands for the corresponding electrical quantity of the inflection point according to the integral of the reduction peak.11,39,40 This is a standard electrochemical method frequently used for determining the ECSA of Pd nanoparticles. | ||
| Fig. 4 CV curves of Pd NPs-modified GCEs in aqueous H2SO4 (1 mol L−1) at a scan rate of 100 mV s−1: Pd-a (a), Pd-b (b), Pd-c (c) and Pd-d (d). | ||
Electrocatalytic activity of Pd NPs for ethanol oxidation
As depicted in Fig. 5A, the Pd NPs showed increased electrocatalytic activity compared to reported Pd catalysts.41 Because the catalytic activity of Pd NPs is mainly dependent on their size,42–44 Among all the Pd NP-modified GCEs, the Pd-a-modified GCE exhibited the highest catalytic current densities (1260 and 1610 mA mg−1 for the positive and negative scans, respectively) during the process of electrooxidation of ethanol; the corresponding values for the Pd-b-modified GCE were 1200 and 1300 mA mg−1, those for the Pd-c-modified GCE were 1070 and 1180 mA mg−1, and those for the Pd-d-modified GCE were 960 and 1100 mA mg−1, respectively. The catalytic activity of the four Pd NP electrocatalysts decreased in the following order: Pd-a > Pd-b > Pd-c > Pd-d. That is, the electrocatalytic activity of ethanol oxidation increased with decreasing particle size, because smaller Pd NPs had an obviously higher ECSA than larger ones. Moreover, as illustrated by the inset of Fig. 5A, the onset potential of the Pd-a modified GCE in the positive direction tended to be obviously more negative than that of Pd-d-modified GCE, indicating that the Pd-a electrocatalyst has advantages with respect to the kinetics of the ethanol oxidation reaction. As shown in Fig. 5B, however, the electrocatalytic activity per real surface area for the Pd-d NPs was much higher than that of the Pd-a NPs. This difference in Fig. 5A might be attributed to the different oxidation rates of ethanol on the (100) and (111) facets of Pd and illustrates the dependence of the electrochemical activity of Pd nanocrystals on the shape. Therefore, the nanocube structure of Pd-d could result in the enhanced adsorptive strength of the CH3CH2O per unit area during alcohol electrooxidation.45 However, the shape effect of Pd nanocrystals on the activity still remains largely unexplored.5,14,15 | ||
| Fig. 5 (A and B) CV curves of Pd NPs-modified GCEs in 1 mol L−1 ethanol + 1 mol L−1 KOH at 50 mV s−1 and (C) chronoamperometric curves of Pd NPs-modified GCEs at 0.3 V: Pd-a (a), Pd-b (b), Pd-c (c) and Pd-d (d). | ||
The catalytic stability data were confirmed by continuous electrochemical measurements. As shown in Fig. 5C, the transient current densities for ethanol oxidation of all the modified electrodes decreased rapidly in the first 40 s and then decreased slowly until 800 s had passed. Finally, a proximate plateau was observed. This might be ascribed to the deactivation of the Pd NPs together with the reduction in solution viscosity and the existence of H+ or OH− ions in the solution.46 In addition, the competitive adsorption of the substrate/partial oxidation product on the catalyst surface caused the reduction of the oxidation reaction rate.46 On the basis of our experimental results, although the alkaline electrolyte could reduce the corrosive action to the metal catalyst during the catalytic process, the palladium catalyst may also gradually lose some activity on account of the corrosive effect.47 Compared with the other three samples, the catalytic current density of the Pd-d-modified electrode remained unchanged after test times over 200 s because the Pd-d NPs owned the most regular shapes among the four samples, and the crystallization nature of the exposed surface was better, which led to its special electrocatalytic stability (Fig. 5C). Above all, it was found that Pd-a possessed the highest electrocatalytic activity and best cyclic stability among all the samples, which can be primarily attributed to the minimum size of the Pd-a NPs.
Fig. 6 shows the cycle stability curves of the modified GCEs for the electrooxidation of ethanol by taking Pd-a and Pd-d as examples. It can be observed that Pd-a exhibited excellent electrocatalytic activity and reached the maximum current density after 25 cycles. Note that approximately 75% of the catalytic activity was maintained through 100 cycles followed by an obvious sustained decrease for the continuous cycles. In the meantime, Pd-d still retained 82% of the maximum catalytic activity after 100 cycles, and the final current density after 200 cycles decreased to 45% of the maximum value; thus, Pd-d showed better catalytic performance than Pd-a. These data were essentially in accordance with the results derived from Fig. 5C.
 | ||
| Fig. 6 Variation in current densities as a function of cycle number for the GCEs modified by Pd-a (a) and Pd-d (b). | ||
The peak potentials of all the modified GCEs would shift in both the positive and negative scans in the process of ethanol oxidation. Taking the performance of Pd-a as an example, the variation in potential with cycle number is shown in Fig. 7. The ladders were the result of intervals when collecting the data recorded by the electrochemical workstation. The peak voltage would shift positively in both the positive and negative scans with increasing circle number; however, the peak voltage of electrocatalytic ethanol became slightly larger in the negative scan, indicating an increase in polarization voltage. The electric current in the negative scan was decreased on account of the palladium oxide that was produced on the surface of palladium during the electrocatalytic process, which would cover up the active site of palladium.48–50 Whereas, the positive shift in the positive scan was caused by the excessive or residual Pd–OHads, which was probably due to the change in the concentration of OH in the solution and then led to the increase in Pd–OHads.
 | ||
| Fig. 7 Relationship between cycle number and peak potential for the positive scan (a) and negative scan (b) of the Pd-a modified GCE. | ||
Conclusions
In summary, we have presented a facile and effective polyol route for the synthesis of Pd NPs with sizes in the range of 3 to 10 nm by using ethylene glycol as the solvent and reduction agent and PVP as the surfactant. It was found that all the Pd NPs were well-crystallized and had narrow size distributions. Experimental data showed that the Pd NPs smaller than 5 nm in size were spherical, and the Pd NPs larger than 6.5 nm in size had distinct shapes. Moreover, the electrocatalytic performance was closely related to the particle size and structure of Pd NPs. The Pd NPs with sizes of 3 nm exhibited the highest catalytic activity and superior stability toward the electrooxidation of ethanol in alkaline solution, which were ascribed to the largest electrochemically active surface area and the minimum size among all the Pd NPs.Acknowledgements
This work was financially supported the National Natural Science Foundation of China (No. 21143006 and U1232104), the Taishan Scholars Advantageous and Distinctive Discipline Program for supporting the research team of energy storage materials of Shandong Province and the Foundation of Taishan Scholar program of Shandong Province, P. R. China.References
- K. H. Young and J. Nei, Materials, 2013, 6, 4574–4608 CrossRef.
- S. B. Yin, M. Cai, C. X. Wang and P. K. Shen, Energy Environ. Sci., 2011, 4, 558–563 CAS.
- S. Y. Shan, J. Luo, J. F. Wu, N. Kang, W. Zhao, H. Cronk, Y. G. Zhao, P. Joseph, V. Petkov and C.-J. Zhao, RSC Adv., 2014, 4, 42654–42669 RSC.
- R. Narayanan and M. A. El-Sayed, Nano Lett., 2004, 4, 1343–1348 CrossRef CAS.
- N. Tian, Z. Y. Zhou and S. G. Sun, Chem. Commun., 2009, 12, 1502–1504 RSC.
- K. M. Bratlie, H. Lee, K. Komvopoulos, P. D. Yang and G. A. Somorjai, Nano Lett., 2007, 12, 3097–3101 CrossRef PubMed.
- M. J. Ren, J. Chen, Y. Li, H. F. Zhang, Z. Q. Zou, X. M. Li and H. Yang, J. Power Sources, 2014, 246, 32–38 CrossRef CAS.
- D. Bosch and E. Pradkhan, Resour. Policy, 2015, 44, 118–134 CrossRef.
- M. Date, M. Okumura, S. Tsubota and M. Haruta, Angew. Chem., Int. Ed., 2004, 43, 2129–2132 CrossRef CAS PubMed.
- H.-C. Chiu and C.-S. Yeh, J. Phys. Chem. C, 2007, 111, 7256–7259 CAS.
- J. Łuczak, M. Paszkiewicz, A. Krukowska, A. Malankowska and A. Zaleska-Medynska, Adv. Colloid Interface Sci., 2016, 230, 13–28 CrossRef PubMed.
- Z. P. Shao, W. Zhou and Z. G. Zhu, Prog. Mater. Sci., 2012, 57, 804–874 CrossRef CAS.
- P. K. Jain, X. Huang, I. H. El-Sayed and M. A. El-Sayed, Acc. Chem. Res., 2008, 41, 1578–1586 CrossRef CAS PubMed.
- B. Lim, M. J. Jiang, J. Tao, P. H. Camargo, Y. M. Zhu and Y. N. Xia, Adv. Funct. Mater., 2009, 19, 189–200 CrossRef CAS.
- X. Q. Huang, S. H. Tang, X. L. Mu, Y. Dai, G. X. Chen, Z. Y. Zhou, F. X. Ruan, Z. L. Yang and N. F. Zheng, Nat. Nanotechnol., 2011, 6, 28–32 CrossRef CAS PubMed.
- Y. J. Xiong, H. G. Cai, B. J. Wiley, J. G. Wang, M. J. Kim and Y. N. Xia, J. Am. Chem. Soc., 2007, 129, 3665–3675 CrossRef CAS PubMed.
- H. Y. Bai, M. Han, Y. Z. Du, J. C. Bao and N. H. Dai, Chem. Commun., 2010, 46, 1739–1741 RSC.
- W.-L. Qu, Z.-B. Wang, Z.-Z. Jiang, D.-M. Gu and G.-P. Yin, RSC Adv., 2012, 2, 344–350 RSC.
- A. J. Armenta-González, R. Carrera-Cerritos, M. Guerra-Balcázar, L. G. Arriaga and J. Ledesma-García, J. Appl. Electrochem., 2015, 45, 33–41 CrossRef.
- M. Chen, Z.-B. Wang, K. Zhou and Y.-Y. Chu, Fuel Cells, 2010, 10, 1171–1175 CrossRef CAS.
- N. V. Longabcde, T. Hayakawab, T. Matsubarab, N. D. Chiend, M. Ohtakie and M. Nogami, J. Exp. Nanosci., 2012, 7, 426–439 CrossRef.
- M. Crespo-Quesada, J.-M. Andanson, A. Yarulin, B. Lim, Y. N. Xia and L. Kiwi-Minsker, Langmuir, 2011, 27, 7909–7916 CrossRef CAS PubMed.
- B. Wiley, Y. G. Sun, B. Mayers and Y. N. Xia, Chem.–Eur. J., 2005, 11, 454–463 CrossRef CAS PubMed.
- Y. J. Xiong, I. Washio, J. Y. Chen, H. G. Cai, Z.-Y. Li and Y. N. Xia, Langmuir, 2006, 22, 8563–8570 CrossRef CAS PubMed.
- Y. G. Sun, B. Mayers, T. Herricks and Y. N. Xia, Nano Lett., 2003, 3, 1171–1175 Search PubMed.
- Y. G. Sun, Y. D. Yin, B. T. Mayers, T. Herricks and Y. N. Xia, J. Mater. Chem., 2002, 14, 4736–4745 CrossRef CAS.
- H. Zhang, M. S. Jin, Y. J. Xiong, B. Lim and Y. N. Xia, Acc. Chem. Res., 2012, 46, 1783–1794 CrossRef PubMed.
- Y. Gao, L. Song, P. Jiang, L. F. Liu, X. Q. Yan, Z. P. Zhou, D. F. Liu, J. X. Wang, H. J. Yuan, Z. X. Zhang, X. W. Zhao, X. Y. Dou, W. Y. Zhou, G. Wang, S. S. Xie, H. Y. Chen and J. Q. Li, J. Cryst. Growth, 2005, 276, 606–612 CrossRef CAS.
- B. Wiley, Y. Sun, J. Shen, H. Cang, Z.-Y. Li and Y. Xia, MRS Bull., 2005, 30, 338–348 CrossRef.
- C. C. Li, R. Sato, M. Kanehara, H. B. Zheng, Y. Bando and T. Teranishi, Angew. Chem., Int. Ed., 2009, 121, 7015–7019 CrossRef.
- T. Gendrineau, S. Marre, M. Vaultier, M. Pucheault and C. Aymonier, Angew. Chem., Int. Ed., 2012, 51, 8525–8528 CrossRef CAS PubMed.
- D. Mandal, K. J. Kim and J. S. Lee, Langmuir, 2012, 28, 10310–10317 CrossRef CAS PubMed.
- R. X. Ding, X. Y. Wu, G. T. Han, Q. C. Wang, H. L. Lu, H. L. Li, A. P. Fu and P. Z. Guo, ChemElectroChem, 2015, 2, 427–433 CrossRef CAS.
- V. Bambagioni, C. Bianchini, J. Filippi, W. Oberhauser, A. Marchionni, F. Vizza, R. Psaro, L. Sordelli, M. L. Foresti and M. Innocenti, ChemElectroChem, 2009, 2, 99–112 CAS.
- Z. Y. Wang, F. P. Hu and P. K. Shen, Electrochem. Commun., 2006, 11, 1764–1768 CrossRef.
- L. Perini, C. Durante, M. Favaro, S. Agnoli, G. Granozzi and A. Gennaro, Appl. Catal., B, 2014, 144, 300–307 CrossRef CAS.
- M. Grdeń, M. Łukaszewski, G. Jerkiewicz and A. Czerwiński, Electrochim. Acta, 2008, 53, 7583–7598 CrossRef.
- M. Grdeń, A. Piascik, Z. Koczorowski and A. Czerwiński, J. Electroanal. Chem., 2002, 532, 35–42 CrossRef.
- P. Z. Guo, Z. B. Wei, W. N. Ye, W. Qin, Q. C. Wang, X. F. Guo, C. J. Lu and X. S. Zhao, Colloids Surf., A, 2012, 395, 75–81 CrossRef CAS.
- S. Song, Y. Wang, P. Tsiakaras and P. K. Shen, Appl. Catal., B, 2008, 78, 381–387 CrossRef CAS.
- Z. P. Li, H. L. Lu, Q. Li, X. S. Zhao and P. Z. Guo, Colloids Surf., A, 2015, 464, 129–133 CrossRef CAS.
- Q. C. Wang, Y. Q. Wang, P. Z. Guo, Q. Li, R. X. Ding, B. Y. Wang, H. L. Li, J. Q. Liu and X. S. Zhao, Langmuir, 2014, 30, 440–446 CrossRef CAS PubMed.
- J. A. Baeza, L. Calvo, M. A. Gilarranz, A. F. Mohedano, J. A. Casas and J. J. Rodriguez, J. Catal., 2012, 293, 85–93 CrossRef CAS.
- X. G. Wang, W. M. Wang, Z. Qi, C. C. Zhao, H. Ji and Z. H. Zhang, J. Power Sources, 2010, 195, 6740–6747 CrossRef CAS.
- I. Park, K. Lee, S. J. Yoo, Y. Cho and Y. Sung, Electrochim. Acta, 2010, 55, 4339–4345 CrossRef CAS.
- C. Bianchini and P. K. Shen, Chem. Rev., 2009, 109, 4183–4206 CrossRef CAS PubMed.
- Z. B. Shao, W. Zhu, H. Wang, Q. H. Yang, S. L. Yang, X. D. Liu and G. Z. Wang, J. Phys. Chem. C, 2013, 117, 14289–14294 CAS.
- F. F. Zhang, D. B. Zhou and M. D. Zhou, J. Nat. Gas Chem., 2016, 25, 71–76 Search PubMed.
- B. Habibi and S. Mohammadyari, Int. J. Hydrogen Energy, 2015, 40, 10833–10846 CrossRef CAS.
- G.-X. Cai, J.-W. Guo, J. Wang and S. Li, J. Power Sources, 2015, 276, 279–290 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1 and S2. See DOI: 10.1039/c6ra18146e |
| This journal is © The Royal Society of Chemistry 2016 |