
Tailored honeycomb-like polymeric films based on amphiphilic poly(urea/malonamide) dendrons†
Abstract
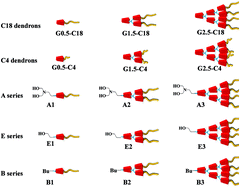
A series of hydrogen bond-rich poly(urea/malonamide) dendrons were utilized as surfactants to facilitate the formation of honeycomb-like porous structures from the breath figure (BF) process. With the addition of a small amount of dendritic surfactants to polymers such as poly(D,L-lactide), polystyrene or poly(methyl methacrylate), a well-organized honeycomb-like surface could be achieved. These uniform porous arrays from the BF method with free-standing capacity benefit from the support of their polymer matrices without resorting to a painstaking polymerization process to provide bulky dendritic side-chain polymers. The formation of water-driven honeycomb-like surfaces was also dependent on the concentration of surfactants in a polymer matrix, apart from the chemical structure. Furthermore, a quantitative analysis of the interfacial tensions between water and polymer solution revealed a dynamic procedure of water droplets stabilized by the surfactants on the as-cast polymer films during the solvent evaporation step of the BF method. Among all these dendritic surfactants, the dendrons with one or two hydroxyl groups at the focal point and plenty of octadecyl groups in the periphery exhibited an amphiphilic nature, and were able to create well-balanced interfacial tensions capable of maintaining water in droplets. Consequently, this type of dendron as a surfactant can be blended with a wide range of polymers to create regular honeycomb-like arrays.
 Please wait while we load your content...
Please wait while we load your content...

