
Facile preparation of a novel SnO2@UiO-66/rGO hybrid with enhanced photocatalytic activity under visible light irradiation†
Abstract
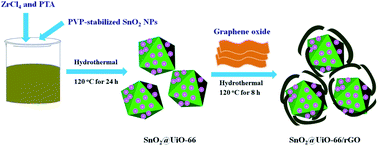
An effective ternary composite of SnO2@UiO-66/rGO was designed and synthesized. The as-prepared samples were characterized by various techniques, including powder X-ray diffraction (XRD), Raman spectroscopy, scanning electron microscopy (SEM), high resolution transmission electron microscopy (HRTEM), N2 adsorption–desorption isotherms, UV-vis diffuse reflection spectroscopy (DRS), zeta potential analysis, photoluminescence (PL) spectroscopy and electrochemical impedance spectroscopy (EIS). The photocatalytic activity of ternary SnO2@UiO-66/rGO, binary SnO2@UiO-66, SnO2@rGO, P25 TiO2 and SnO2 were studied by employing the degradation of rhodamine B (RhB) from water under visible light irradiation. The results showed that the degradation efficiency of SnO2@UiO-66/rGO was found to be 95.5% for RhB dyes within 150 min, which is much superior than the others. The enhanced photocatalytic property of the ternary composite can be attributed to the microporous structure, high surface area, enhanced visible light absorption, efficient charge transfer process, as well as the synergetic effect among SnO2 NPs, UiO-66 and rGO. Therefore, these kinds of MOF-based ternary composites have great potentiality in environmental remediation fields.
 Please wait while we load your content...
Please wait while we load your content...

