
Hybrid bicelles as a pH-sensitive nanocarrier for hydrophobic drug delivery†
Abstract
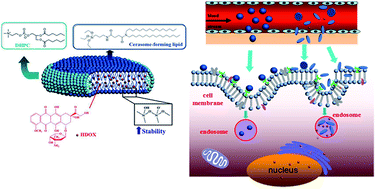
Hydrophobic doxorubicin was successfully loaded into hybrid discoid bicelles generated from proamphiphilic organoalkoxysilane and dihexanoyl phosphatidylcholine at the ratio of 7 : 2 by conventional Bangham method in combination with sol–gel reaction and self-assembly process. The drug-loaded hybrid bicelles with about 60 nm diameter and 6 nm thickness were found to exhibit pH-sensitive release behavior, good biocompatibility and remarkably high stability towards surfactant solubilization, long-term storage, and many factors susceptible to destabilize conventional phospholipid bicelles. The hybrid bicelles were proved to have higher cellular uptake via endocytosis and adhesion than spherical cerasomes. The endocytosis of hybrid bicelles was related to clathrin, macropinocytosis and was energy-dependent. Both in vitro and in vivo results showed that the drug loaded bicelles can effectively inhibit tumor growth. In other words, such hybrid bicelles can be employed as a novel promising nanocarrier for hydrophobic drugs.
 Please wait while we load your content...
Please wait while we load your content...

