
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gadolinium-doped magnetite nanoparticles from a single-source precursor†
F. J. Douglasa,
D. A. MacLaren*b,
N. Macleana,
I. Andreu c,
F. J. Kettlesa,
F. Tunad,
C. C. Berrye,
M. Castroc and
M. Murrie*a
c,
F. J. Kettlesa,
F. Tunad,
C. C. Berrye,
M. Castroc and
M. Murrie*a
aWestCHEM, School of Chemistry, University of Glasgow, Glasgow G12 8QQ, UK. E-mail: Mark.Murrie@glasgow.ac.uk; Tel: +44 141 330 4486
bSUPA, School of Physics and Astronomy, The University of Glasgow, Glasgow G12 8QQ, UK. E-mail: dmaclaren@physics.org; Tel: +44 141 330 5886
cInstituto de Ciencia de Materiales de Aragón (ICMA), CSIC – Universidad de Zaragoza, Campus Río Ebro, María de Luna, 3, 50018 Zaragoza, Spain
dNational EPR Centre, University of Manchester, Oxford Road, Manchester, M13 9PL, UK
eCentre for Cell Engineering, CMVLS, University of Glasgow, Glasgow G12 8QQ, UK
First published on 27th July 2016
Abstract
An iron and gadolinium-containing bimetallic polynuclear complex was used as a single source precursor in the synthesis of gadolinium-doped magnetite nanoparticles (Gd:Fe3O4). The synthesis produces well defined octahedral particles (12.6 ± 2.6 nm diameter) with a gadolinium content in the region of 2 mol%. The nanoparticles showed a value of the specific absorption rate of 3.7 ± 0.6 W gFe−1 under low-amplitude radiofrequency magnetic field excitation, and moderate biocompatibility, suggesting that these particles are viable candidates for magnetic hyperthermia applications.
Introduction
The last decade has seen a surge in interest in the synthesis and characterisation of nanoparticles (NPs), driven by their unique properties and their potential uses across a range of applications. Magnetic nanoparticles (MNPs) are amongst the most intensively studied, and in medical applications have both therapeutic and diagnostic prospects, as agents for magnetic hyperthermia (MH),1 drug delivery and magnetic resonance imaging (MRI) contrast.2,3 However, creating MNPs that are ‘tuned’ for specific purposes is far from trivial and careful refinement of synthetic procedures is essential to gain precise control over the particle morphology, composition and magnetic properties including the saturation magnetisation, remnant magnetisation and coercivity.4–7 Much of the literature focuses on metal oxides, particularly magnetite, because of its ease of production, stability and biocompatibility – some iron oxide nanospecies are already approved by the FDA and the European Commission for clinical applications8,9 and have been used in clinical trials.10,11An interesting prospect is to further enhance the properties of MNPs through doping, such as the use of low levels of Zn2+ doping in ferrite particles to enhance their MRI and MH effect.12 Here, we focus on the composite material gadolinium-doped magnetite (Gd:Fe3O4). Gd doping or labelling has been shown to improve the MRI contrast of iron oxide NPs via T1 enhancement,13–17 whereas the MH properties of gadolinium-doped magnetite have received much less attention.1,18 Whilst doping ions into MNPs is simple in principle, establishing reproducible synthetic procedures can be difficult. For example, co-precipitation or simultaneous decomposition protocols require the metal precursors to react/decompose with similar kinetics if both metals are to be incorporated within a single product.19 Differences in reactivity could lead to inhomogeneous products or complete segregation into two discrete nanoparticle populations, a problem that motivates the present report. Our own initial attempts to control the simultaneous decomposition of separate iron- and gadolinium-containing precursors were unreliable, often resulting in distinct nucleation of two NP species.20 For this reason we elected to synthesise and use a novel single-source precursor, a bimetallic iron/gadolinium complex. This is advantageous in nanoparticle synthesis because decomposition of the precursor is guaranteed to release iron and gadolinium ions into the reaction mixture simultaneously, which should reduce separate nucleation and low dopant ion uptake. A further advantage of this protocol is that polynuclear precursors can also provide an extra degree of freedom with which to optimise a reaction.5
Experimental
Materials and methods
GdCl3·2H2O (99%), FeCl2·4H2O (99%), sodium benzoate (99%), benzyl ether (98%), oleic acid (90%), oleyl amine (70%), acetonitrile (MeCN) and diethyl ether (Et2O) were purchased from Sigma Aldrich and used as received without further purification.Synthesis of the Fe/Gd precursor
The synthesis was adapted from that used to prepare [Fe2CuO(O2CPh)6(H2O)3].21 A solution of sodium benzoate (1.88 g, 13 mmol) in H2O (100 mL) was added to a stirred solution of FeCl2·4H2O (0.5 g, 2.5 mmol) and GdCl3·2H2O (0.46 g, 1.25 mmol) in H2O (50 mL). MeCN (25 mL) was then added to this solution. The reaction mixture was stirred overnight at ambient temperature to give a light brown precipitate. This precipitate was collected by filtration and washed with Et2O (see ESI†).Synthesis of Gd:Fe3O4 NPs
0.2 g of the Fe/Gd air-dried precursor was added to a reaction mixture consisting of oleylamine![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :oleic acid:benzyl ether in the ratio 4.5:3:2.5 mL. The mixture was then heated to 110 °C under a nitrogen flow for 30 minutes. The nitrogen flow was reduced and the mixture was slowly heated to 310 °C for 30 minutes. The mixture was then cooled to room temperature and the particles were precipitated with excess ethanol and collected via centrifugation as a black/brown residue and redispersed in hexane.
:oleic acid:benzyl ether in the ratio 4.5:3:2.5 mL. The mixture was then heated to 110 °C under a nitrogen flow for 30 minutes. The nitrogen flow was reduced and the mixture was slowly heated to 310 °C for 30 minutes. The mixture was then cooled to room temperature and the particles were precipitated with excess ethanol and collected via centrifugation as a black/brown residue and redispersed in hexane.
Transmission electron microscopy and elemental analysis
Samples were prepared for Transmission Electron Microscopy (TEM) characterisation by dispersing a small amount of dilute hexane particle dispersion directly onto a holey-carbon coated copper TEM grid. TEM and scanning TEM (STEM) were performed on a Tecnai TF20 operated at 200 kV. The microscope was fitted with an EDAX Energy dispersive X-ray spectroscopy (EDX) system and a Gatan Enfina Spectrometer for Electron Energy Loss Spectroscopy (EELS) measurements. TEM data were obtained and processed using either Digital Micrograph or IMAGEJ 1.41 software.Magnetic measurements
Magnetic measurements were performed on the oleate-protected NPs using a Quantum Design MPMS-XL7 SQUID magnetometer. Samples were fixed in eicosane in gelatine capsules prior to measurement. Data have been corrected for the contributions of the sample holder and eicosane matrix.Aqueous phase transfer
To test the potential of the Gd:Fe3O4 NPs for biological applications, the hydrophobic oleate surfactant of the as-synthesized particles was replaced with a water-dispersible PEG derivative ligand (PEG600-DPA) as described elsewhere.5,22Cell culture
Human dermal fibroblasts cell line h-TERT BJ1 were cultured and expanded in DMEM/10% FBS (100 U mL−1 penicillin, 1000 μg mL−1 streptomycin) and maintained at 37 °C in 5% CO2 until ∼90% confluent. Cells were seeded at a density of 1 × 104 cells per ml. Prior to incubation, particles suspensions were filter sterilised (0.2 μ filter) and diluted in fresh media to the required concentration.MTT assay
To assess the toxicity of the NPs, the cell metabolic activity was determined via an MTT assay. Particle suspensions at the required concentrations were incubated with h-TERT BJ1 cells in a 96 well plate for 1 hour at 37 °C. The particle suspension was then removed and 5 μL of MTT dye (5 mg mL−1 in phosphate buffer pH 7.4, Sigma-Aldrich) was added to each well. After 1.5 hours of incubation at 37 °C, the medium was removed and any formazan crystals produced were dissolved in 100 μL of DMSO. The absorbance of each well was read on a microplate reader (Dynatech MR7000 instruments) at 550 nm, calibrated to zero absorbance using culture medium without cells. Details of the cell culture protocol and additional electron microscopy imaging of cells with the NPs can be found in the ESI.†Specific absorption rate (SAR) measurement
The sample for the SAR measurement was prepared starting from an aqueous dispersion of the PEG functionalized Gd:Fe3O4 NPs.22 A small amount of the aqueous dispersion was placed inside a polycarbonate capsule. Evaporation of the water was accelerated by gentle heating until a viscous (due to the PEG content) dry sample was obtained and no changes to the specimen weight (sample + capsule) were detected (±0.01 mg) over one day. The total specimen weight was 31 mg with a sample mass (PEG 600-DPA functionalized Gd:Fe3O4 nanoparticles) of 0.82 mg.The ability of the NPs to generate heat under external radio frequency (RF) excitation was determined by adiabatic magnetothermia.23 The specimen, kept under adiabatic conditions, was subjected to an alternating magnetic field with frequency f = 111 kHz and amplitude H0 = 3 kA m−1 (38 Oe) during a time interval Δt of approximately 5 minutes. The temperature increase during the magnetic field pulse, ΔT, was measured and the performance of the sample was characterized through its Specific Absorption Rate (SAR), calculated as:
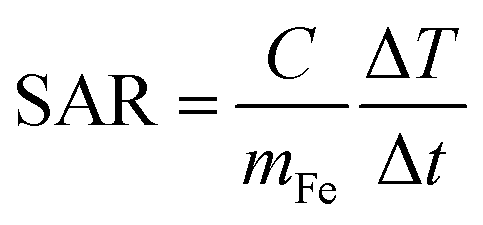 | (1) |
Results and discussion
Gd:Fe3O4 NP synthesis and characterisation
TEM images of a typical sample of NPs are presented in Fig. 1. The particles are well-defined and octahedral, with a measured average long axis length of 12.6 ± 2.6 nm (Fig. 1a). The size and shape of nanoparticles is determined in part by the adsorption of molecular species during synthesis, particularly if binding affinities vary with the crystallographic orientation of exposed surfaces.4 Octahedral or truncated octahedral Fe3O4 nanoparticles are known to arise from the competitive adsorption of oleate and oleylamine species,24 the relative concentrations of which determine the precise NP shape.25 The NP shape can also be perturbed by the presence of other molecules, which includes the decomposition products of the molecular precursor used here.5 High resolution TEM (Fig. 1b) confirmed that the particles are single crystalline, with lattice fringes clearly visible and with the fringe spacings consistent with those of magnetite. The inverse spinel crystal structure typical of Fe3O4 was confirmed via selected area electron diffraction (SAED, Fig. 1c), suggesting that Gd3+ uptake does not have a significant effect on overall crystal structure. A STEM EDX line trace across a single particle and a composite EDX spectrum obtained from a group of particles are shown in Fig. 1d and e and clearly support the presence of both iron and gadolinium in the particles. There is no clear variation in the Gd:Fe ratio across the particle, nor contrast variation in STEM images that would suggest Gd segregation or a core–shell morphology and we conclude that the Gd is uniformly distributed throughout the magnetite lattice. | ||
| Fig. 1 TEM analysis of the Gd:Fe3O4 particles, showing (a) an overview of the NPs, with a clear prevalence of octahedral particles. (b) High resolution TEM image of a typical octahedral particle, annotated with fringe spacings that are consistent with those of magnetite. (c) SAED, with diffraction rings indexed with those of the magnetite (Fe3O4) structure. (d) Iron and gadolinium Lα signal intensities from a STEM EDX line trace across a single particle (see inset dark-field image), showing the co-location of Fe and Gd in a single NP. (e) EDX spectrum obtained from a group of particles, showing the presence of iron and gadolinium. The Cu peaks arise from secondary scattering from the TEM support grid. | ||
STEM-EELS measurements were also performed and have the advantage that the obtained spectra provide an accurate representation of the chemical composition of a material at a given point without, for example, the secondary effects that produce the spurious Cu signal in EDX. This spatially-resolved compositional analysis is critical in ruling out the presence of inhomogeneous products and to definitively demonstrate Gd incorporation within the particles. To minimise electron beam damage, data was acquired by ‘hopping’ the electron beam from particle to particle, dwelling on each particle for 5 seconds. The particles chosen for this measurement are shown in Fig. 2a. The EELS spectra obtained from roughly 35 particles is presented in Fig. 2b–d, which shows well defined edges for oxygen-K and iron-L2,3 edges, in addition to a signal from the gadolinium-M5,4 edges. The calculated elemental composition was determined to be Fe: 47.6%, O: 50.8% and Gd: 1.6%, using standard background-fitting, removal of plural scattering and EELS cross-sections within the Gatan Digital Micrograph software.26 Subsequent EELS measurements taken from a larger number of samples across the grid indicate a variation in fitted Gd content from 1.6% to 2.5%, giving a mean Gd content for the MNP sample of around 2 (±0.4) mol%.
 | ||
| Fig. 2 (a) Dark-field STEM image of typical Gd:Fe3O4 NPs used for EELS analysis (in dark field images, heavy material such as the nanoparticles appear bright). (b–d) Background-subtracted EELS edges, showing the presence of O, Fe and Gd in the particles. | ||
When compared with other methods for producing Gd:Fe3O4 particles, our single source high-temperature method has distinct advantages. For example, a previous mild co-precipitation method between iron and gadolinium chlorides at 65 °C produced ill-defined particles with a single-domain maximum gadolinium level of ∼1 mol%.27 The authors were able to increase the Gd3+ levels to a maximum of 1.4 mol%, though the resultant particles were polydisperse, aggregated and believed to be multidomain. It should be noted that the co-precipitation method does allow for some control over the level of Gd3+ dopant,28 though this is often at the expense of control over size distribution or particle morphology, which is retained in our thermal decomposition method. In contrast, our single source method produced well defined NPs with a gadolinium content in the range 2 (±0.4) mol%. The observed gadolinium levels for our single source particles are in agreement with levels seen elsewhere in the literature. Wang et al. reported a maximum Gd3+ dopant level of 2.85 mol% in cobalt-ferrite microparticles.29 Above this level, the nucleation of a separate GdFeO3 perovskite phase was observed, suggesting an upper limit to Gd incorporation within the magnetite lattice of a few mol%. This is not surprising given the difference in ionic radii between Gd3+ (107.8 pm), Fe2+ (78 pm) and Fe3+ (65 pm).30
The SAED measurements of Fig. 1c confirm that the magnetite structure has been retained upon gadolinium doping. We found no evidence for the formation of other gadolinium ferrite phases, such as perovskite GdFeO3 or garnet Gd3Fe5O12.31 The observed lattice parameter of 8.21 Å is consistent with that of magnetite and may reflect the fact that there are two competing trends in determining the lattice parameter: a reduction in lattice parameter due to relaxation effects within small NPs and lattice expansion due to gadolinium incorporation.29 It is possible that the incorporation of Gd may inhibit the growth of larger particles, due to increasing difficulties in accommodating lattice strain. Despite thorough experimentation, we were unable to obtain larger particles by altering the reaction conditions – implying that the 12.6 nm particles obtained here may represent the upper grain size limit using these reaction conditions. However, we note that previous work on the Gd:Fe3O4 system yielded particles up to 33 nm with increasing Gd3+ content, albeit with an increase in polydispersity and above ∼25 nm the particles became multi-domain.27
Magnetic measurements were performed to investigate the effect of magnetic ion doping on the magnetisation of the doped magnetite particles. Hysteresis loops recorded at 100 K and 300 K are provided in Fig. 3a. Field-cooled (FC) and zero field-cooled (ZFC) measurements were also performed and are shown in Fig. 3b. The hysteresis loops clearly show the superparamagnetic behaviour of the particles. Interestingly, the magnetisation (M) values for the Gd:Fe3O4 particles (32.9 and 28.9 emu g−1 at 100 and 300 K respectively) were lower than the 96.4 emu g−1 expected for bulk magnetite at room temperature.32 This follows similar trends for lanthanide doped magnetite, as Drake et al. reported room temperature M values of 65.7 emu g−1 for superparamagnetic Gd0.02Fe2.98O4 NPs, synthesised by the co-precipitation method.1 Liang et al. also prepared lanthanide-doped magnetite particles using the co-precipitation method. Of these the gadolinium-doped particles showed the highest room temperature M values of 45.9 emu g−1.33 De Silva et al. published a high temperature thermolysis route to Ln:Fe3O4 (Ln = Sm, Eu, and Gd) particles.34 In this report the M values for Sm:Fe3O4 and Eu:Fe3O4 particles are 42.1/31.3 and 29.8/23.6 emu g−1 (Sm and Eu particles at 60 K and 300 K, respectively), though no comment is made regarding the M values of the Gd:Fe3O4 particles. This decrease seems to be not limited to the low Gd3+ doping level since it has been also determined for GdxFe3−xO4 NPs (average size 8 nm) obtained by standard chemical precipitation process for the compositional range 0.1 < x < 1.9.35 Decreasing M values with increasing levels of Ln3+ doping have also been observed for other ferrite systems, such as gadolinium doped in γ-Fe2O3 powders,36 cobalt-ferrite microparticles,29 and Cu0.5Zn0.5Fe2−xLnxO4 powders, where a decrease in magnetisation upon Ln3+ doping (increasing x from 0 to 0.02) was observed.34
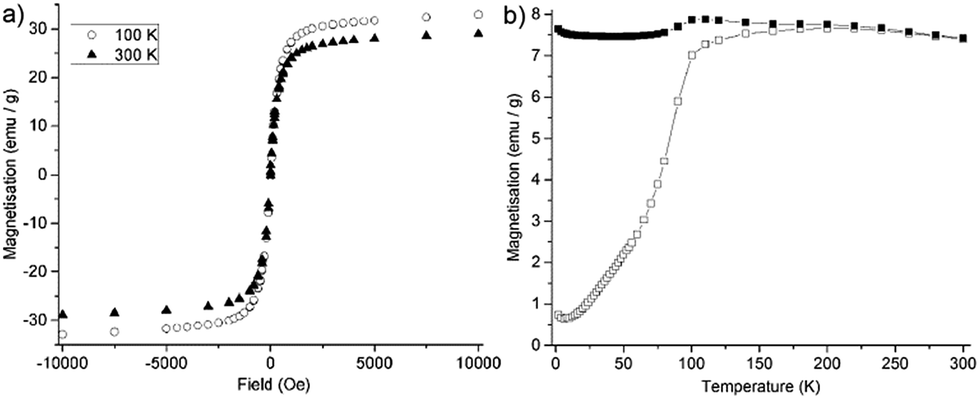 | ||
| Fig. 3 (a) Hysteresis loops for Gd:Fe3O4 nanoparticles recorded at 100 and 300 K. (b) Zero field cooled (open symbols) and field-cooled (closed symbols) magnetisation of octahedral 12 nm Gd:Fe3O4 particles in an applied field of 100 Oe. | ||
The most likely explanation for the observed decrease in magnetisation upon doping is that Ln3+ ions cause a disruption to ferrimagnetic ordering.1 The difference in ionic radii between gadolinium(III) and iron(II)/(III) ions is in the order of 30 pm (for Fe2+) and 43 pm (for Fe3+). In lanthanide-doped magnetite, the large Gd3+ ion will go into the octahedral B sites of the lattice, increasing strain in the lattice and therefore reducing the overall crystalline symmetry, which will increase the magnetocrystalline anisotropy.29,37 Reduction of the overall crystalline symmetry may also disrupt the ferrimagnetic ordering within the magnetite lattice, which would contribute to the lower overall magnetisation of the particle. The decrease in magnetisation seen in our Gd:Fe3O4 NPs may therefore appear to be the consequence of two factors: a decrease in long range magnetic ordering upon insertion of large Gd3+ ions into the Fe3O4 matrix and an increase in magnetic anisotropy. This is an encouraging result, as Gd doping provides a way of modifying the anisotropy of the particles.
Field cooled (FC) and zero field cooled measurements (ZFC) were recorded for the Gd:Fe3O4 NPs, and the data are presented in Fig. 3b, from which a blocking temperature (TB) of ∼200 K was extracted. The measured TB is in agreement with previously reports; for example, Park et al. reported TB values of ∼110 K and ∼225 K for 12 and 16 nm particles respectively.6 The TB is directly related to the magnetic anisotropy and the volume of the particles. However, it is strongly affected by the effect of magnetic interactions,38 and therefore the TB values do not contradict the aforementioned increase in magnetic anisotropy.
Turning to MH measurements, the hydrophobic oleylamine/oleic acid surfactant layer protecting the as-prepared particles was first exchanged with a hydrophilic PEG derivative in order to facilitate aqueous dispersion of the particles. Successful surfactant exchange was confirmed by an increase in the hydrodynamic radius and by the evident stability of the PEG coated particles in aqueous dispersion (see Fig. S3†). The PEG coated particles were then tested for cell uptake and toxicity using human fibroblast cells. The MTT assay showed that cell metabolic activity (viability) remained in the 70–80% region for Gd:Fe3O4 particles for concentrations below 0.1 mg mL−1 and in the 90–100% region for undoped Fe3O4 particles (see Fig. S4†). The reasons behind the slight decrease in cell viability in the gadolinium doped samples are unclear; it may be possible that there is ‘leakage’ of free Gd3+ from the doped particles which is in turn having a detrimental effect on cell viability. This would have to be improved before potential clinical applications of Gd:Fe3O4 nanoparticles could be realised,39 with further studies to improve the surface coating needed. Optical fluorescence microscopy and electron microscopy were performed on the cells incubated with both doped and undoped particles (see Fig. S5 and S6† respectively). Fluorescence microscopy shows a change in phenotype, with a more rounded cell shape, and cytoskeletal reorganisation on incubation with the particles, most likely due to cellular uptake (Fig. S5†), whilst electron microscopy clearly demonstrated an increase in cell membrane activity and particle uptake into cells (using SEM and TEM, respectively, Fig. S6†).40
The specific absorption rate (SAR) of the PEG-coated NPs was determined at room temperature by adiabatic magnetothermia.23 As the specimen heat capacity, C, is derived only from the polycarbonate capsule (the estimated sample contribution is lower than 5% and can be considered as negligible), the calculated SAR value is 3.7 ± 0.6 W gFe−1.
There is little literature about SAR measurements of Gd3+ doped iron oxide NPs for comparison. An intriguing SAR enhancement with Gd3+ doping respect to pure magnetite has been reported for GdxFe3−xO4 with x = 0.01–0.03 which increases as the Gd doping increases.1,27 However, size effects cannot be ruled out since the particle diameter also increases, from 13 to 19 nm, with the Gd3+ doping level. SAR also depends strongly on the alternating magnetic field parameters. While our measurements were performed under low field amplitude H0 = 3 kA m−1 and f = 111 kHz, the SAR values around 30–40 W gFe−1 reported in ref. 1 and 23 were measured under H0 = 246 Oe (=19.5 kA m−1) and f = 52 kHz. The dependence of SAR on the alternating field parameters depends on the size, polydispersity, anisotropy constant and the strength of the magnetic interactions, and cannot be predicted a priori.41,42 However, as a rule of thumb, a square (linear) dependence with H0(f) can be considered, assuming the linear response theory and a non-interacting nanoparticle assembly.43 The extrapolation of the experimental values in the literature1,27 to our experimental alternating magnetic field parameters gives a SAR of 1.5–2 W gFe−1. Hence, the sample studied in the present work, with a higher Gd3+ content, obtained using a single-source precursor shows an improved SAR value with respect to previously reported values. We postulate that the effect of the Gd3+ dopant on the magnetic anisotropy constant is the reason for such enhancement.
Conclusion
A bimetallic iron gadolinium single-source precursor was used to synthesise high quality gadolinium-doped magnetite (Gd:Fe3O4) NPs by a surfactant-assisted thermolysis route. We have shown that polynuclear complexes can be used as precursors in the synthesis of MNPs and that mixed metal complexes provide a facile route to obtain doped particles, which can be otherwise difficult to prepare. TEM measurements showed that ∼12 nm diameter octahedral particles were formed, with EDX and EELS measurements confirming the presence of gadolinium in the particles in the region of 2 ± 0.4 mol%. SQUID measurements revealed that the presence of gadolinium ions in the particles is responsible for a decrease in the magnetisation. This could be accounted for by a change in the magneto-crystalline structure of the magnetite lattice due to the doping of the large Gd3+ ions, which will induce a change in the magneto-crystalline anisotropy. Biological studies show that cells exposed to Gd:Fe3O4 NPs do not react adversely and MH measurements showed the particles to have a relatively high SAR value of 3.7 ± 0.6 W gFe−1 (when H0 = 3 kA m−1 and f = 111 kHz).Acknowledgements
We thank Dr Emma Humphreys-Williams at the Imaging and Analysis Centre (IAC), Natural History Museum, London for MNP metal content analysis. FJD gratefully acknowledges a scholarship from the University of Glasgow and DAM a fellowship from the UK Engineering and Physical Sciences Research Council (grant ref. EP/I00419X/1). IA was supported by a JAE-Predoc 2010 (CSIC program “Junta para la ampliación de estudios”), co-funded by the European Social Fund. We also acknowledge financial support by the Spanish MEC (MAT2011-24284) and MINECO (Project MAT2013-44063-R).References
- P. Drake, H.-J. Cho, P.-S. Shih, C.-H. Kao, K.-F. Lee, C.-H. Kuo, X.-Z. Lin and Y.-J. Lin, J. Mater. Chem., 2007, 17, 4914–4918 RSC.
- K. Krishnan, IEEE Trans. Magn., 2010, 46, 2523–2558 CrossRef CAS PubMed.
- A. Hervault and N. T.-K. Thanh, Nanoscale, 2014, 6, 11553–11573 RSC.
- F. J. Douglas, D. A. MacLaren and M. Murrie, RSC Adv., 2012, 2, 8027–8035 RSC.
- F. J. Douglas, D. A. MacLaren, F. Tuna, W. M. Holmes, C. C. Berry and M. Murrie, Nanoscale, 2014, 6, 172–176 RSC.
- J. Park, K. An, Y. Hwang, J. Park, H. Noh, J. Kim, J. Park, N. Hwang and T. Hyeon, Nat. Mater., 2004, 3, 891–895 CrossRef CAS PubMed.
- W. S. Seo, H. H. Jo, K. Lee, B. Kim, S. J. Oh and J. T. Park, Angew. Chem., Int. Ed., 2004, 43, 1521–3773 CrossRef.
- A. L. Cortajarena, D. Ortega, S. M. Ocampo, A. González-García, P. Couleaud, R. Miranda, C. Belda-Iniesta and A. Ayuso-Sacido, Nanobiomedicine, 2014, 1, 1–20 CrossRef.
- L. Li, W. Jiang, K. Luo, H. Song, F. Lang, Y. Wu and Z. Gu, Theranostics, 2013, 3, 595–615 CrossRef PubMed.
- K. Maier-Hauff, R. Rothe, R. Scholz, U. Gneveckow, B. Thiesen, A. Feussner, A. Deimling, N. Waldoefner, R. Felix and A. Jordan, J. Neuro-Oncol., 2007, 81, 53–60 CrossRef CAS PubMed.
- K. Maier-Hauff, F. Ulrich, D. Nestler, H. Niehoff, P. Wust, B. Thiesen, H. Orawa, V. Budach and A. Jordan, J. Neuro-Oncol., 2011, 103, 317–324 CrossRef PubMed.
- J.-T. Jang, H. Nah, J.-H. Lee, S. H. Moon, M. G. Kim and J. Cheon, Angew. Chem., Int. Ed., 2009, 48, 1234–1238 CrossRef CAS PubMed.
- K. H. Bae, Y. B. Kim, Y. Lee, J. Hwang, H. Park and T. Gwan Park, Bioconjugate Chem., 2010, 21, 505–512 CrossRef CAS PubMed.
- Z. Zhou, L. Wang, X. Chi, J. Bao, L. Yang, W. Zhao, Z. Chen, X. Wang, X. Chen and J. Gao, ACS Nano, 2013, 7, 3287–3296 CrossRef CAS PubMed.
- T.-H. Shin, Y. Choi, S. Kim and J. Cheon, Chem. Soc. Rev., 2015, 44, 4501–4516 RSC.
- N. Xiao, W. Gu, H. Wang, Y. Deng, X. Shi and L. Ye, J. Colloid Interface Sci., 2014, 417, 159–165 CrossRef CAS PubMed.
- X. Wang, Z. Zhou, Z. Wang, Y. Xue, Y. Zeng, J. Gao, L. Zhu, X. Zhang, G. Liu and X. Chen, Nanoscale, 2013, 5, 8098–8104 RSC.
- E. Roy, S. Patra, R. Madhuri and P. K. Sharma, Colloids Surf., B, 2016, 142, 248–258 CrossRef CAS PubMed.
- S. Chen, D. A. MacLaren, R. T. Baker, J. N. Chapman, S. Lee, D. J. Cole-Hamilton and P. André, J. Mater. Chem., 2011, 21, 3646–3654 RSC.
- F. J. Douglas, PhD Thesis, University of Glasgow, 2012 Search PubMed.
- A. Ferguson, J. McGregor, E. K. Brechin, L. H. Thomas and M. Murrie, Dalton Trans., 2009, 9395–9397 RSC.
- J. Xie, C. Xu, N. Kohler, Y. Hou and S. Sun, Adv. Mater., 2007, 19, 3163–3166 CrossRef CAS.
- E. Natividad, M. Castro and A. Mediano, Appl. Phys. Lett., 2008, 92, 093116 CrossRef.
- L. Zhang, J. Wu, H. Liao, Y. Hou and S. Gao, Chem. Commun., 2009, 4378–4380 RSC.
- Q. Song, Y. Ding, Z. L. Wang and Z. J. Zhang, J. Phys. Chem. B, 2006, 110, 25547–25550 CrossRef CAS PubMed.
- Gatan Inc. website, http://www.gatan.com/, accessed Feburary 2016.
- P.-S. Jiang, P. Drake, H.-J. Cho, C.-H. Kao, K.-F. Lee, C.-H. Kuo, X.-Z. Lin and Y.-J. Lin, J. Nanosci. Nanotechnol., 2012, 12, 5076–5081 CrossRef CAS PubMed.
- G. Zhang, R. Du, L. Zhang, D. Cai, X. Sun, Y. Zhou, J. Zhou, J. Qian, K. Zhong, K. Zheng, D. Kaigler, W. Liu, X. Zhang, D. Zou and Z. Wu, Adv. Funct. Mater., 2015, 25, 6101–6111 CrossRef CAS.
- Q. Wang, S. Li, A. Wu and H. Yang, J. Magn. Magn. Mater., 2009, 321, 2622–2626 CrossRef CAS.
- C. E. Housecroft and A. G. Sharpe, Inorganic Chemistry, Pearson Education, 2001 Search PubMed.
- F. Söderlind, L. Selegârd, P. Nordblad, K. Uvdal and P.-O. Käll, J. Sol-Gel Sci. Technol., 2009, 49, 253–259 CrossRef.
- A. Angermann and J. Topfer, J. Mater. Sci., 2008, 43, 5123–5130 CrossRef CAS.
- X. Liang, X. Wang, J. Zhuang, Y. Chen, D. Wang and Y. Li, Adv. Funct. Mater., 2006, 16, 1805–1813 CrossRef CAS.
- C. R. D. Silva, S. Smith, I. Shim, J. Pyun, T. Gutu, J. Jiao and Z. Zheng, J. Am. Chem. Soc., 2009, 131, 6336–6337 CrossRef PubMed.
- V. N. Nikoforov and B. L. Oksengendler, Inorg. Mater., 2014, 50, 1222–1225 CrossRef CAS.
- A. Nikumbh, J. Mater. Sci., 1990, 25, 3773–3779 CrossRef CAS.
- Y.-I. Kim, W. B. Im, M. K. Jeon, Y.-H. Lee, K.-B. Kim and K.-S. Ryu, J. Nanosci. Nanotechnol., 2011, 11, 810–814 CrossRef CAS PubMed.
- I. Andreu, L. Solozabal, E. Natividad and O. Roubeau, ACS Nano, 2015, 9, 1408–1419 CrossRef CAS PubMed.
- J. G. Penfield and R. F. Reilly, Nat. Rev. Nephrol., 2007, 3, 654–668 Search PubMed.
- C. C. Berry, J. Mater. Chem., 2005, 15, 543–547 RSC.
- J. Carrey, B. Mehdaoui and M. Respaud, J. Appl. Phys., 2011, 109, 083921 CrossRef.
- D. Serantes, D. Baldomir, C. Martinez-Boubeta, K. Simeonidis, M. Angelakeris, E. Natividad, M. Castro, A. Mediano, D.-X. Chen, A. Sanchez, L. I. Balcells and B. Martínez, J. Appl. Phys., 2010, 108, 073918 CrossRef.
- R. E. Rosensweig, J. Magn. Magn. Mater., 2002, 252, 370–374 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, additional microscopy images. See DOI: 10.1039/c6ra18095g |
| This journal is © The Royal Society of Chemistry 2016 |