
Role of flexible bulky groups and weak interactions involving halogens in the vapoluminescence of a metal-free dye†
Abstract
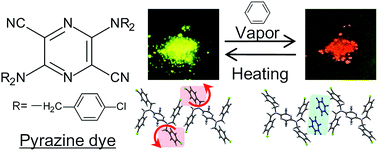
A metal-free pyrazine dye exhibited vapoluminescence when exposed to benzene vapour. Single-crystal X-ray analysis revealed that its flexible bulky groups and weak interactions involving halogens play an essential role in the occurrence of the observed vapoluminescence.
 Please wait while we load your content...
Please wait while we load your content...

