
High-performance dielectric ionic ladderphane-derived triblock copolymer with a unique self-assembled nanostructure†
Jie Chen,
Dandan Zhou,
Cuifang Wang,
Xiaojuan Liao,
Meiran Xie* and
Ruyi Sun*
School of Chemistry and Molecular Engineering, East China Normal University, Shanghai 200241, China. E-mail: mrxie@chem.ecnu.edu.cn; rysun@chem.ecnu.edu.cn
First published on 12th September 2016
Abstract
Functional poly(bisnorbornene)-based ladderphanes, P1, P2, and P3, as well as the derived triblock copolymers containing two blocks of poly(N-3,5-difluorophenyl-norbornene pyrrolidine) (PFNP), PFNP-b-P2-b-PFNP and PFNP-b-P3-b-PFNP, were synthesized by ring-opening metathesis polymerization. The dielectric constants of the polymers were 5, 7, 21, 12, and 18 accompanied with the dielectric losses of 0.03, 0.03, 0.11, 0.02, and 0.04 at a frequency range of 100 Hz to 1 MHz, respectively. In particular, the ionic copolymer PFNP-b-P3-b-PFNP could self-assemble into a unique tree ring-like nanostructure, in which the ionic P3 blocks were isolated between the insulating PFNP blocks, and the long distance migration of ions was difficult to achieve, resulting in lower dielectric dissipation and higher charge–discharge efficiency than those of the ionic homopolymer P3. This research presented a practical way to effectively improve the dielectric properties by combining the ionic, dipolar, and nano-interfacial polarizations, as well as the stereoregular microstructure of the polymer chain.
Introduction
High-performance dielectric materials play an important role in advanced electrical and power applications.1,2 Compared with conventional ceramic counterparts,3–6 polymer-based dielectric materials have some advantages7,8 such as a light weight, low cost, higher breakdown strength, and lower dielectric loss, which therefore make them an ideal choice for energy storage, film capacitor, and pulsed power applications.2,5,9,10 However, the limiting factor for large polymer-based capacitors is the low permittivity (2–5) and energy density of polymers.3 The dielectric performance of polymers is usually affected by the electronic, orientational (or dipolar), ionic, and interfacial polarizations.11 Therefore, combining multiple polarization is envisioned to enhance the relative dielectric constant and also effectively increase the energy storage (Ue = 0.5kε0E2)11 of dielectric polymers, while it is still a challenge to gain excellent dielectric properties of polymers. As a result, the dielectric percolative composites are widely used as energy storage materials depending on the interfacial polarization between the conductive fillers (e.g. metal particles) and insulating polymer matrix.12–14 Recently, plenty of new structures and conductive fillers,15 including polyaniline,16 polythiophene,17 graphene,18 and carbon nanotubes,19 are used with the expectation of improving the dispensability of the conductive segments, enhancing the compatibility of the composite components, and ultimately decreasing the dielectric dissipation and the frequency dependency.Polymerized ionic liquids and ionic polymers have a relatively high density of strong dipoles,4,20 and the dipoles tend to prefer parallel alignment to avoid the strongly overlap of polarizability volumes, effectively.4 So they can offer relatively high static dielectric constant. Besides, the dielectric loss is not very dependent on frequency under 1000 Hz.21 However, the hysteresis that accompanies the transport of ions species over a long distance exhibited a high dielectric loss.21 So, introducing ionic group as a strategy to prepare high-permittivity thin-film capacitor is hard to accomplish because of high dielectric loss. However, a multilayer or core–shell nanoarchitecture strategy can effectively decrease dielectric loss from ion migration in polar polymers because ionic polymers are isolated between the insulating blocks.15,22,23 Meanwhile, the covalent-linkage between the conductive and insulating moieties can also be beneficial to improve the compatibility and reach a low dielectric loss.3,12
Polynorbornene (PNBE) and perylene bisimide (PBI) are usually known as low-dielectric materials,24–26 and they are used in the microelectronics industry9,24 and organic photovoltaic materials,27–33 respectively. Ladderphane, a new class of double-stranded polymers,34,35 has greater resistance to irradiation as well as thermal and chemical degradation in comparison to their single-stranded counterparts. PNBE can readily be functionalized by the introduction of conjugated aromatic structure and ionic groups to greatly increase the dielectric constant.4,20,36–38 Besides, with the stereoregular chain configuration and cofacial PBI linkers, the accumulation of dipole moments9 and electron-delocalization39,40 can also promote the dielectric properties. Therefore, combining multiple polarization and constructing a unique nanostructure may enhance the dielectric constant of ionic polymers while keeping a low dielectric loss. In this article, we report the synthesis of ladderphane structure-based ionic triblock copolymer by ring-opening metathesis polymerization (ROMP) via a simple process, and these copolymers could self-assemble into a tree ring-like nanoarchitecture, which endowed the ionic copolymers excellent dielectric properties.
Experimental
Materials
endo-N-(4-Hydroxy phenyl)-norbornene-dicarboximide (endo-NDI), carboxyl-contained perylene bisimide (PBI-COOH), N-3,5-difluorophenyl-norbornene-pyrrolidine (FNP), and bis(norbornene dicarboximide) with a PBI linker (1) were prepared as the previous procedures.34 [1,3-Bis(2,4,6-trimethylphenyl)-4,5-dihydroimidazol-2-ylidene][3-bromo-pyridine]2benzylidene ruthenium dichloride (Ru-III) was prepared according to the literature.41 1,6,7,12-Tetrachloroperylene-3,4:9,10-tetracarboxylic dianhydride was purchased from commercial sources at analytical grade and used without further purification. 12-Amino dodecanoic acid was obtained from Alfa Aesar. 3,6-endo-Methylene-1,2,3,6-tetrahydrophthalic anhydride, lithium aluminum hydride (LiAlH4) were purchased from Aldrich or Alfa Aesar and used as received. Methyl iodide, potassium hexafluorophosphate (KPF6), 4-dimethylaminopyridine (DMAP), acryloyl chloride, ethyl acetate (EtOAc), petroleum ether, magnesium sulfate (MgSO4), glacial acid (HAc), and propionic acid were purchased from Shanghai Chemical Reagents Co, and used as received without purification. Solvents were distilled over drying agents under nitrogen prior to use: dichloromethane (CH2Cl2), trichloromethane (CHCl3) from calcium hydride; tetrahydrofuran (THF) from sodium/benzophenone. 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (EDCI·HCl), 4-dimethylaminopyridine (DMAP), 2-aminoethanol, and 2-ethylhexylamine were purchased from Energy Chemical. All reactions were carried out under dry nitrogen atmospheres using standard Schlenk-line techniques.Characterization
1H (500 MHz) and 13C (125 MHz) NMR spectra were recorded using tetramethylsilane as an internal standard on a Bruker DPX spectrometer. Melting point was determined by X-4B apparatus. IR spectra were recorded by Perkin Elmer Spectrum One FTIR spectrophotometer. Elemental analysis was conducted with an Elementar vario EL. Gel permeation chromatography (GPC) was used to calculate relative molecular weight and molecular weight distribution equipped with a Waters 1515 Isocratic HPLC pump, a Waters 2414 refractive index detector, and a set of Waters Styragel columns (7.8 × 300 mm, 5 mm bead size; 103, 104, and 105 Å pore size). GPC measurements were carried out at 40 °C using 0.05 mol L−1 bis(trifluoromethane)sulfonimide lithium salt-contained THF as the eluent with a flow rate of 1.0 mL min−1. The system was calibrated with polystyrene standard. Thermal gravimetric analysis (TGA) was performed using an SDTA851e/SF/1100 TGA instrument under nitrogen flow at a heating rate of 10 °C min−1 from 25 to 800 °C. Differential scanning calorimeter (DSC) was performed on a Netzsch 204F1 in nitrogen atmosphere. An indium standard was used for temperature and enthalpy calibrations. All the samples were first heated from 40 to 250 °C and held at this temperature for 3 min to eliminate the thermal history, and then, they were cooled to room temperature and heated again from 40 to 250 °C at a heating or cooling rate of 10 °C min−1. Transmission electron microscopy (TEM) images were recorded on the JEOL2100F microscopes operating at 120 kV. Samples for TEM measurement were prepared by depositing a drop of THF solution with different concentration on the copper grids coated with carbon, followed by air-drying. Additionally, the samples were not stained before measurement because the electron density difference between the two blocks provided sufficient contrast for TEM imaging. The hydrodynamic diameter was determined by means of dynamic light scattering (DLS) analysis using a Malvern Zetasizer Nano-ZS light scattering apparatus (Malvern Instruments, U.K.) with a He–Ne laser (633 nm, 4 mW). The Nano ZS instrument incorporates noninvasive backscattering (NIBS) optics with a detection angle of 173°. The z-average diameter of the sample was automatically provided by the instrument using cumulate analysis. Fluorescence measurements were performed on a RF 5302 Shimadzu spectrofluorometer (Japan) equipped with a xenon light source (UXL-150S, Ushio, Japan). The emission and excitation slit widths were 5 nm and 5 nm, respectively. The samples were excited at 530 nm and emission spectra were recorded from 510 to 750 nm. The emission fluorescence values at 545–549 nm was used for subsequent calculations. The critical micelle concentration (CMC) was determined by plotting the wave length correlate to concentration of copolymers. The CMC was taken as the intersection of regression lines calculated from the linear portions of the plot. Dielectric measurements were carried out by a Novocontrol BDS40 dielectric spectrum analyzer over the frequency range of 100 Hz to 1 MHz at room temperature, and a contacting electrode method was used. The edge side (d) of the guarded electrode is 0.3 mm. The capacitance (Cp) and the dissipation factor (dielectric loss, or tan![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) δ) of the tested films were recorded. The dielectric constant (εr) can be calculated by eqn (1):
δ) of the tested films were recorded. The dielectric constant (εr) can be calculated by eqn (1):| εr = Cpl/εoA | (1) |
000 V with a cycle frequency of 1000 Hz.
General procedure for polymerization
Typically, polymerization was carried out in a Schlenk tube under dry nitrogen atmosphere at 30 °C in CHCl3 for a preset time. After confirmed the monomer conversion by TLC, ethyl vinyl ether (0.2 mL) was added to the reaction mixture and stirred for 1 h, and the mixture was concentrated and poured into an excess of acetone. The dark red polymer was washed with acetone, and dried in a vacuum oven at 40 °C to a constant weight.200 and 1.4, respectively. 1H NMR (500 MHz, CF3COOD, ppm): δ 8.95–8.70 (br, pery), 7.88–7.31 (br, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH on phenyl), 5.81–5.42 (br, trans-CH on backbone), 4.55–4.12 (br, CH2NCO), 4.00–3.79 (br, CH2NAr), 3.78–2.95 (br, CHCHCH + CHCHCH + NCH3), 2.86–2.56 (br, OCOCH2), 1.97–0.96 (br, CHCHCH2 + OCOCH2(CH2)9CH2N). 13C NMR (125 MHz, CF3COOD, ppm): δ 172.91, 162.19, 146.32, 141.33, 135.27, 132.89, 131.41, 128.53, 123.53, 121.25, 113.40, 53.26, 50.51, 40.76, 34.33, 30.95, 29.14, 28.91, 27.89, 26.59, 24.88. IR (KBr): 2933 (CH2), 2850 (C C), 1756 (CO), 1707, 1589, 1515, 1392, 1362, 1288, 1238, 1204, 1170, 1101, 970, and 810 cm−1.800 and 1.5, respectively. 1H NMR (500 MHz, CDCl3, ppm): δ 8.77–8.50 (br, pery), 7.44–6.72 (br, CHCH on phenyl), 6.24–5.95 (br, CH on m-difluorophenyl), 5.54–5.20 (br, trans-CH on backbone), 4.35–4.04 (br, CH2NCO), 3.82–3.52 (br, CH2NAr), 3.33–2.67 (br, CHCHCH + CHCHCH + NCH3), 2.66–2.37 (br, OCOCH2), 1.96–0.72 (br, CHCHCH2 + OCOCH2(CH2)9CH2N). 13C NMR (125 MHz, CDCl3, ppm): δ 172.82, 165.21, 164.04, 161.81, 138.29, 135.35, 133.07, 131.28, 128.34, 123.06, 121.89, 113.11, 95.54, 55.73, 50.49, 46.38, 41.11, 34.09, 31.15, 28.81, 28.21, 27.65, 26.48, 24.08. IR (KBr): 2928 (CH2), 2850 (CC), 1756 (CO), 1707, 1667, 1633, 1584, 1510, 1475, 1392, 1362, 1288, 1234, 1209, 1165, 1106, 972, and 815 cm−1.
CH on phenyl), 5.81–5.42 (br, trans-CH on backbone), 4.55–4.12 (br, CH2NCO), 4.00–3.79 (br, CH2NAr), 3.78–2.95 (br, CHCHCH + CHCHCH + NCH3), 2.86–2.56 (br, OCOCH2), 1.97–0.96 (br, CHCHCH2 + OCOCH2(CH2)9CH2N). 13C NMR (125 MHz, CF3COOD, ppm): δ 172.91, 162.19, 146.32, 141.33, 135.27, 132.89, 131.41, 128.53, 123.53, 121.25, 113.40, 53.26, 50.51, 40.76, 34.33, 30.95, 29.14, 28.91, 27.89, 26.59, 24.88. IR (KBr): 2933 (CH2), 2850 (C C), 1756 (CO), 1707, 1589, 1515, 1392, 1362, 1288, 1238, 1204, 1170, 1101, 970, and 810 cm−1.800 and 1.5, respectively. 1H NMR (500 MHz, CDCl3, ppm): δ 8.77–8.50 (br, pery), 7.44–6.72 (br, CHCH on phenyl), 6.24–5.95 (br, CH on m-difluorophenyl), 5.54–5.20 (br, trans-CH on backbone), 4.35–4.04 (br, CH2NCO), 3.82–3.52 (br, CH2NAr), 3.33–2.67 (br, CHCHCH + CHCHCH + NCH3), 2.66–2.37 (br, OCOCH2), 1.96–0.72 (br, CHCHCH2 + OCOCH2(CH2)9CH2N). 13C NMR (125 MHz, CDCl3, ppm): δ 172.82, 165.21, 164.04, 161.81, 138.29, 135.35, 133.07, 131.28, 128.34, 123.06, 121.89, 113.11, 95.54, 55.73, 50.49, 46.38, 41.11, 34.09, 31.15, 28.81, 28.21, 27.65, 26.48, 24.08. IR (KBr): 2928 (CH2), 2850 (CC), 1756 (CO), 1707, 1667, 1633, 1584, 1510, 1475, 1392, 1362, 1288, 1234, 1209, 1165, 1106, 972, and 815 cm−1.Results and discussion
ROMP of bisnorbornene monomers affording poly(bisnorbornene)-based ladderphanes and their block copolymers
With the obtained functional monomers 1, 2, and 3 in hand (ESI†), the PBI-bridged homopolymer ladderphanes, P1, P2, and P3, as well as the triblock copolymer ladderphanes containing two blocks of poly(N-3,5-difluorophenyl-norbornene pyrrolidine) (PFNP), PFNP-b-P2-b-PFNP and PFNP-b-P3-b-PFNP, were subsequently prepared by the third-generation Grubbs catalyst (Ru-III)-initiated ROMP. The polymer structures are shown in Scheme 1. As our previous reported procedure,34 the PBI-linked ladderphane P125 was obtained from 1 under a feed ratio ([M]/[Cat]) of 25 in CHCl3, which had the molecular weight (Mn) of 48.3 kDa and polydispersity index (PDI) of 1.6 (Run 1, Table 1). Similarly, 2 could be converted into the PBI-linked ladderphane P225 completely in half an hour at the same lower feed ratio, having Mn of 45.8 kDa and PDI of 1.5 (Run 2). By increasing the loading of 2, the higher [M]/[Cat] ratios of 50 and 100 (Runs 3 and 4) were attempted for expecting to obtain polymers with higher Mn, however, the conversions of 2 were reduced to 73% and 60%, respectively, even if the reaction time was extended to 3 h. The unreacted monomers were proved to exist in the reaction system by GPC analysis (Fig. S5, ESI†), and the corresponding polymers displayed the Mn values of 57.0 and 80.7 kDa, which were unproportionate to the increased [M]/[Cat] ratios. The ladderphane P225 could be easily soluble in common organic solvents such as CH2Cl2 and CHCl3. | ||
| Scheme 1 Structure of polymers. | ||
| Run | Polymer | t (h) | [M]:[FNP]:[Cat]b |
Mnc (kDa) | PDIc | Yield (%) |
|---|---|---|---|---|---|---|
| a Polymerization conditions: using Ru-III as catalyst, CHCl3 as solvent, T = 30 °C, [M] = 6 × 10−3 mol L−1 for ladderphane polymers.b The molar ratios in feed of monomers to initiator for homo- and copolymerizations.c Determined by GPC in THF relative to monodispersed polystyrene standards.d Polymerization conditions: [M] = 2 × 10−3 mol L−1 in CH2Cl2. | ||||||
| 1 | P125 | 0.5 | 25:0:1 |
48.3 | 1.6 | 98 |
| 2 | P225 | 0.5 | 25:0:1 |
45.8 | 1.5 | 99 |
| 3 | P250 | 3 | 50:0:1 |
57.0 | 1.5 | 73 |
| 4 | P2100 | 3 | 100:0:1 |
80.7 | 1.6 | 60 |
| 5 | PFNP50-b-P225-b-PFNP50 | 3 | 25:50:1 |
81.5 | 1.4 | 95 |
| 6d | P325 | 0.5 | 25:0:1 |
58.2 | 1.4 | 95 |
| 7d | PFNP50-b-P325-b-PFNP50 | 3 | 25:50:1 |
102.8 | 1.5 | 97 |
| 8d | PFNP100-b-P325-b-PFNP100 | 3 | 25:100:1 |
119.5 | 1.5 | 85 |
| 9d | PFNP200-b-P325-b-PFNP200 | 3 | 25:200:1 |
136.6 | 1.5 | 72 |
In order to gain block copolymers, a low [2]/[Cat] ratio of 25 was adopted to guarantee the conversion of 2 nearly 100% in a relatively short period of time (0.5 h). After completed ROMP of 2, the second monomer FNP at the [FNP]/[Cat] ratio of 50 (Run 5) was added to this orange-red mixture of living P2, copolymerization of FNP was then run for 2.5 h. Because each ladderphane P2 had two propagating carbene ends, the corresponding triblock copolymer (PFNP50-b-P225-b-PFNP50) with Mn of 81.5 kDa in 95% yield was finally obtained, which has an obvious advantage of ladderphane structure for the synthesis of ABA triblock copolymer through the relatively simple process. The characteristics for polymerization and the resultant polymers were recorded in Table 1.
For monomer 3, following the previous reaction conditions, ROMP was performed with a molar feed ratio of 3 to Ru-III ([M]/[Cat]) of 25:1 in CHCl3. At the first trial, a large number of precipitation was observed when the concentration of 3 is at 6 × 10−3 mol L−1 at 30 °C, and the reaction mixture gradually transformed from orange solution to orange solid in 0.5 h. So it was considered to choose a good solvent of CH2Cl2 and reduce the concentration of 3 to 2 × 10−3 mol L−1 to enable ROMP conducted in a homogeneous process. With a [M]/[Cat] ratio of 25, nearly 100% of 3 was converted into the PBI-linked ladderphane P325 with Mn of 58.2 kDa and relatively narrow PDI of 1.4. It should be noted that the solubility played an important role in solution characterization of polymer and solution-processed nanostructures. As the poor solubility of P325 was concerned, the triblock copolymer contained more soluble PFNP was synthesized by ROMP to ensure the solubility and optimize the dielectric properties of copolymers. Similar result with PFNP50-b-P225-b-PFNP50 was found for the synthesis of triblock copolymer PFNP50-b-P325-b-PFNP50 in CH2Cl2, when the molar ratios of [3]:[FNP]:[Cat] were 25:50:1 (Run 7), the resultant polymer had Mn of 102.8 kDa in 97% yield. As increasing FNP loading, the polymerization under higher [FNP]/[Cat] ratios of 100 and 200 (Runs 8 and 9) were attempted for expecting to obtain triblock copolymers with the longer insulating PFNP blocks so as to improve the dielectric properties, and the Mn values of PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 increased to 119.5 and 136.6 kDa in 85% and 72% yields, respectively. The ionic triblock copolymers could be soluble in common organic solvents such as CH2Cl2, CHCl3, and THF (Table S1, ESI†).
Structure of ladderphanes
The 1H NMR spectra of P225 and P325 (Fig. 1a and b) showed that the signal of olefinic protons on the norbornene ring at about 6.36 ppm was completely disappeared after ring opening, and no residual olefinic proton signals due to the unreacted norbornene moiety were observed, whereas the new signals of olefinic protons on the backbone came at 5.55–5.30 ppm and 5.81–5.42 ppm, respectively, which pointed out the polymerization was processed successfully. Based on the previous studies,9,42 the trans/cis double bond of as-synthesized polymers could be analyzed by the NMR spectroscopy, and the single signals at around 5.6 ppm in 1H NMR and at 131–133 ppm in 13C NMR (Fig. S6a and b, ESI†) related to the vinyl group suggested that all-trans P225 and P325 formed. | ||
| Fig. 1 1H NMR spectra of P225 (a), P325 (b, in CF3COOD), PFNP50-b-P225-b-PFNP50 (c), and PFNP50-b-P325-b-PFNP50 (d). | ||
In the IR spectra (Fig. S7, ESI†), P225 and P325 showed a solely absorption band at 966 cm−1 for the trans double bond but no appearance at 720 cm−1 for the cis double bond,10 and the observation was in agreement with the NMR analyses. These results are contrast to that the PNBE chain in P125 has mixed trans and cis double bonds,34 as shown in Scheme S2,† because the size of each dicarboximide segment (6.5 Å) in PNBE was greater than 5.0 Å,43 the repulsion between the adjacent carbonyl group greatly impedes the regular microstructure of PNBE formed from endo-monomers during the polymerization process. In order to reduce the repulsion, the neighboring repeat NBE units could form a cis/trans mixed configuration. Fortunately, the size of rigid pyrrolidine moiety was 2.5 Å, which was helpful to tailor high-trans configuration in backbone comparing with dicarboximide structure,9 which was beneficial to improve the dielectric constant.
For representative copolymers PFNP50-b-P225-b-PFNP50 and PFNP50-b-P325-b-PFNP50, the ladderphane P2 or P3/PFNP block ratio could be analyzed by integration of the 1H NMR signals (Fig. 1c and d) related to the aromatic protons (Hk) on PBI linker at 8.75–8.55 and 8.77–8.50 ppm and those on m-difluorobenzene group (Ho+p) at 6.21–5.95 and 6.24–5.95 ppm, and it was found to be nearly 2:3, suggesting that the ladderphane/PFNP block ratio is closed to 1:2, which was almost consistent with the molar ratio in feed for the preparation of copolymers. However, because of the overlap of the signals from the phenyl group on the chain end with those from the aryl group in the ladderphane block, the molecular weight of block copolymers could not be calculated from the 1H NMR spectra by the end-group analysis.
The 13C NMR spectroscopy is an effective means to characterize the microstructure of polymers. In the case of endo-type substituted PNBE derivatives, the signals of methylene carbon can provide an evidence to judge the stereoregularity of polymer chain. There were four different microstructures containing two conformations syn/anti if the tacticity was isotactic or syndiotactic, suggesting that the orientation of side groups attached to polymer chain.9,10 Therefore, the peaks of methylene carbon (Cc) would be clear as in one or two sets of signals. As shown in 13C NMR spectra (Fig. S6, ESI†), although the signals of methylene (Cc) and methyne carbon (Cd) were overlap, the highly trans P225 and PFNP50-b-P225-b-PFNP50 showed only one set of wide peak in the methylene carbon at 40.5–41.5 ppm, indicating that the main chain had a high stereoregularity because there was no multiple peaks of methylene carbon. Similarly, the highly trans P325 and ionic P3 blocks of PFNP50-b-P325-b-PFNP50 adopted isotactic stereochemistry showing two sets of signals in methylene carbon at 38.5–41.0 ppm, and the counterpart of Cc in non-ionic PFNP blocks of PFNP50-b-P325-b-PFNP50 with a high stereoregularity presented one set of wide signal at 40.5–41.5 ppm because the signals of methylene (Cc) and methyne carbon (Cd) were overlap. The existence of rigid pyrrolidine moiety has a positive contribution to form the tactic polymer chain during ROMP process.
Thermal properties of ladderphanes
The thermal stability of polymers was investigated by means of thermal gravimetric analysis (TGA), and the TGA experiments are run from 30 to 800 °C at a heating rate of 20 °C min−1 under nitrogen. As shown in Fig. 2a, the thermal decomposition temperatures (Td, 5% weight loss) of P225, P325, PFNP50-b-P225-b-PFNP50, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200 were 390, 360, 342, 270, 288, and 312 °C sequentially. When the polymers were heated to 800 °C, the residues remain more than 30%. The thermal resistance for ionic P325 overall decreases in comparison to homopolymer P225, which was likely due to the basicity of the counter-anion in P325, which could catalyze decomposition.44 The triblock copolymer PFNP50-b-P225-b-PFNP50 has two weight loss processes at around 307 and 377 °C, which was attributed to the pyrolysis of the different copolymer chains. An initial thermal degradation about 307 °C was assigned to the loss of PFNP blocks, and the next thermal degradation above 377 °C was assigned to the degradation of ladderphane P2 blocks. This indicated that the ladderphane structure played an important role on increasing the thermal stability of polymer. Similarly, ionic copolymer PFNP50-b-P325-b-PFNP50 exhibited two stage decomposition attributed to the pyrolysis of linear PFNP blocks (270–361 °C) and ionic ladderphane P3 blocks (above 361 °C), respectively. Due to the basicity of the counter-anion, the Td of PFNP50-b-P325-b-PFNP50 was lower than the corresponding non-ionic copolymer. Besides, ionic copolymers PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 had a higher initial thermal degradation than PFNP50-b-P325-b-PFNP50 because of the high Mn of PFNP blocks. | ||
| Fig. 2 TGA (a) and DSC (b) curves of polymers. | ||
The DSC technique was invited to obtain the glass transition temperature (Tg) and all the samples are first heated from 40 to 250 °C and hold at this temperature for 3 min to eliminate the thermal history, and then, they were cooled to room temperature and heat again from 40 to 250 °C at a heating or cooling rate of 10 °C min−1. From the DSC curves (Fig. 2b), the high Tgs for P225, P325, PFNP50-b-P225-b-PFNP50, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200 were observed at 184, 215, 178, 203, 205, and 209 °C sequentially, which were much higher than those of corresponding homopolymers and their conventional copolymers, likely due to the rigid ladderphane structure.9,10,12 As for P325, since it tends to packing orderly between molecular chains by ionic interaction,45 the motion of backbone was restricted, leading to an increased temperature to attain the relaxation process and an elevated Tg. Importantly, the high Tg could prevent the dielectric loss from electronic and ionic conductions, which could improve the dielectric properties.11 Besides, neither the crystallization peak nor the melting peak could be observed during the cooling (Fig. S8, ESI†) or heating process.
Self-assembly behavior of ladderphanes
The self-assembly of polymers was verified by dynamic light scattering (DLS), which showed that the aggregates existed in solution with different number-average hydrodynamic diameter (Dh) in the selective solvent of THF. As shown in Fig. 3, there was only one peak with a Dh of 13 nm corresponding to a single molecule of the non-ionic homopolymer P225 at a diluted concentration of 0.005 mg mL−1 and no aggregates were observed in THF solution; while the ionic homopolymer P325 displayed two peaks with the Dhs of 8 nm (6.0%) and 58 nm (94.0%), which proved the coexistence of the single polymer chain and the assembly of P325 in THF at the same low concentration likely due to the attraction between cation and anion. For the non-ionic copolymer PFNP50-b-P225-b-PFNP50, although the rigid P2 block and the flexible PFNP block were dissolved in THF well, the disparity of stiffness between the constituent blocks resulted in an increase in the Flory–Huggins χ-parameter,46 so the self-assembly behavior of PFNP50-b-P225-b-PFNP50 could occur, affording two distinct peaks with a dominating Dh of 57 nm (75.1%) for the assembly and a little small Dh of 12 nm (24.9%) for the single polymer chain at 0.005 mg mL−1 in THF. As the concentration increased to 0.01 mg mL−1, the dominating Dh moved to 92 nm (84.3%) and another small Dh of 17 nm (15.7%) still existed. When the concentration was at 0.02 mg mL−1, the dominating Dh and another Dh were further up to 152 nm (88.7%) and 19 nm (11.3%), respectively. Obviously, the mean volume of single polymer chain with small Dh decreased gradually with the increase of concentration, accompanied by the more and larger aggregates.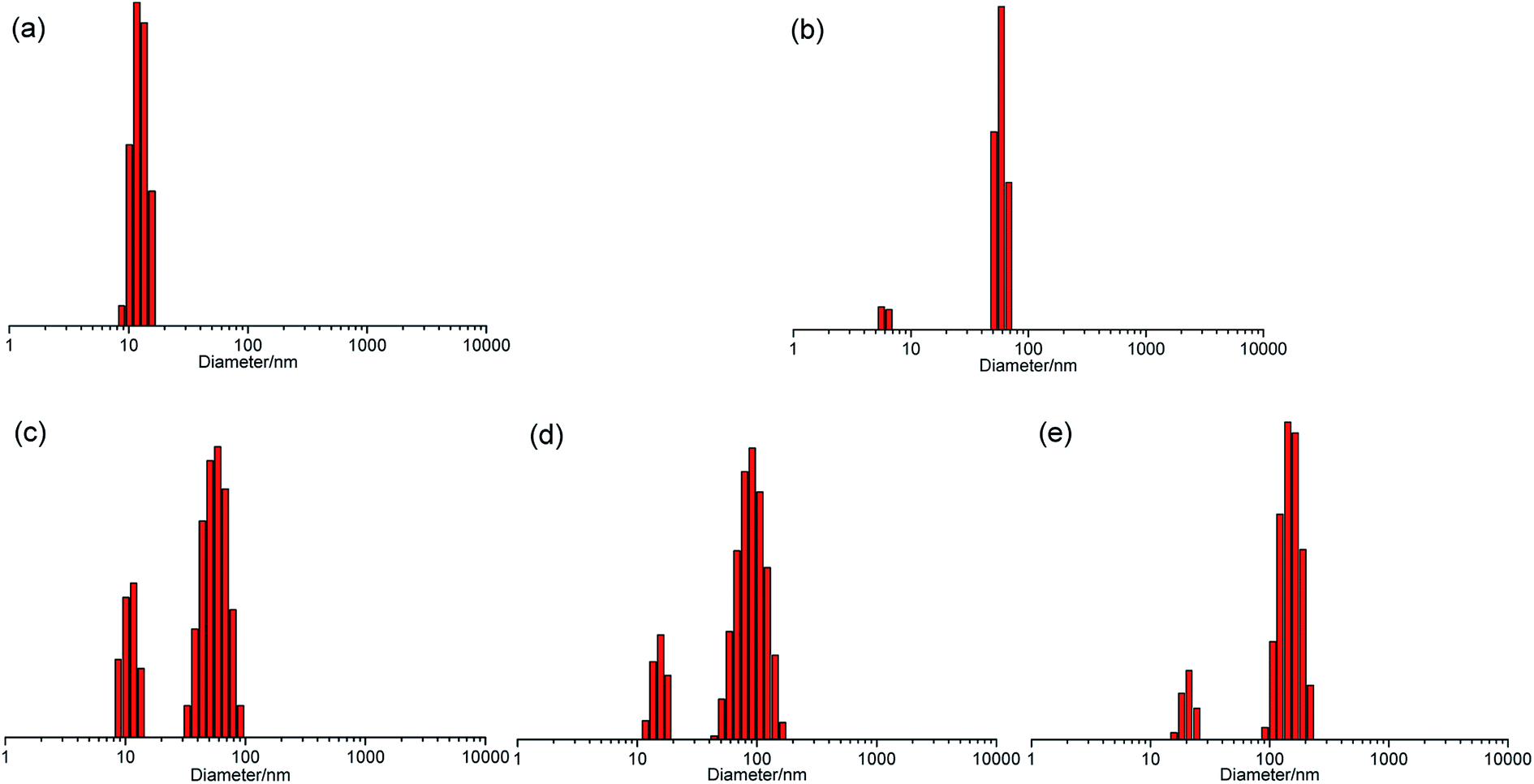 | ||
| Fig. 3 Sizes of P225 (a), P325 (b), and PFNP50-b-P225-b-PFNP50 (c–e) at 0.005 (a–c), 0.01 (d), and 0.02 (e) mg mL−1 in THF determined by means of DLS. | ||
In contrast, the ionic copolymer PFNP50-b-P325-b-PFNP50 formed only one peak for the self-assembled nanostructure with a Dh of 53 nm at 0.005 mg mL−1 in THF (Fig. 4), and the Dhs moved to 66 and 74 nm as the concentration increased to 0.01 and 0.02 mg mL−1, respectively, which dictated that the self-assembled nanostructures grew with the increase of concentration. Similarly, the Dhs of PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 were 73 and 79 nm at 0.005 mg mL−1 in THF. With the concentration increased to 0.01 and 0.02 mg mL−1, the Dhs increased to 85 and 87 nm, as well as 110 and 134 nm, respectively. The DLS analysis showed that the ionic copolymers were easy to obtain the self-assembled nanostructures than the non-ionic copolymers.
 | ||
| Fig. 4 Sizes of PFNP50-b-P325-b-PFNP50 (a–c), PFNP100-b-P325-b-PFNP100 (d–f), and PFNP200-b-P325-b-PFNP200 (g–i) at 0.005 (a, d and g), 0.01 (b, e and h), and 0.02 (c, f and i) mg mL−1 in THF determined by means of DLS. | ||
The CMC was a key factor to determine the self-assembly of polymers. For block copolymers PFNP50-b-P225-b-PFNP50, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200, the CMC values were calculated to be 2.4 × 10−3, 8.9 × 10−4, 6.3 × 10−4, and 5.8 × 10−4 g L−1, respectively (Fig. S9, ESI†). The CMC values of non-ionic copolymer PFNP50-b-P225-b-PFNP50 was greater than the corresponding ionic copolymer PFNP50-b-P325-b-PFNP50, which was matched well with their self-assembly ability as depicted in DLS analysis. For ionic copolymers, the CMC values were gradually decreased with the increase of molecular weight.
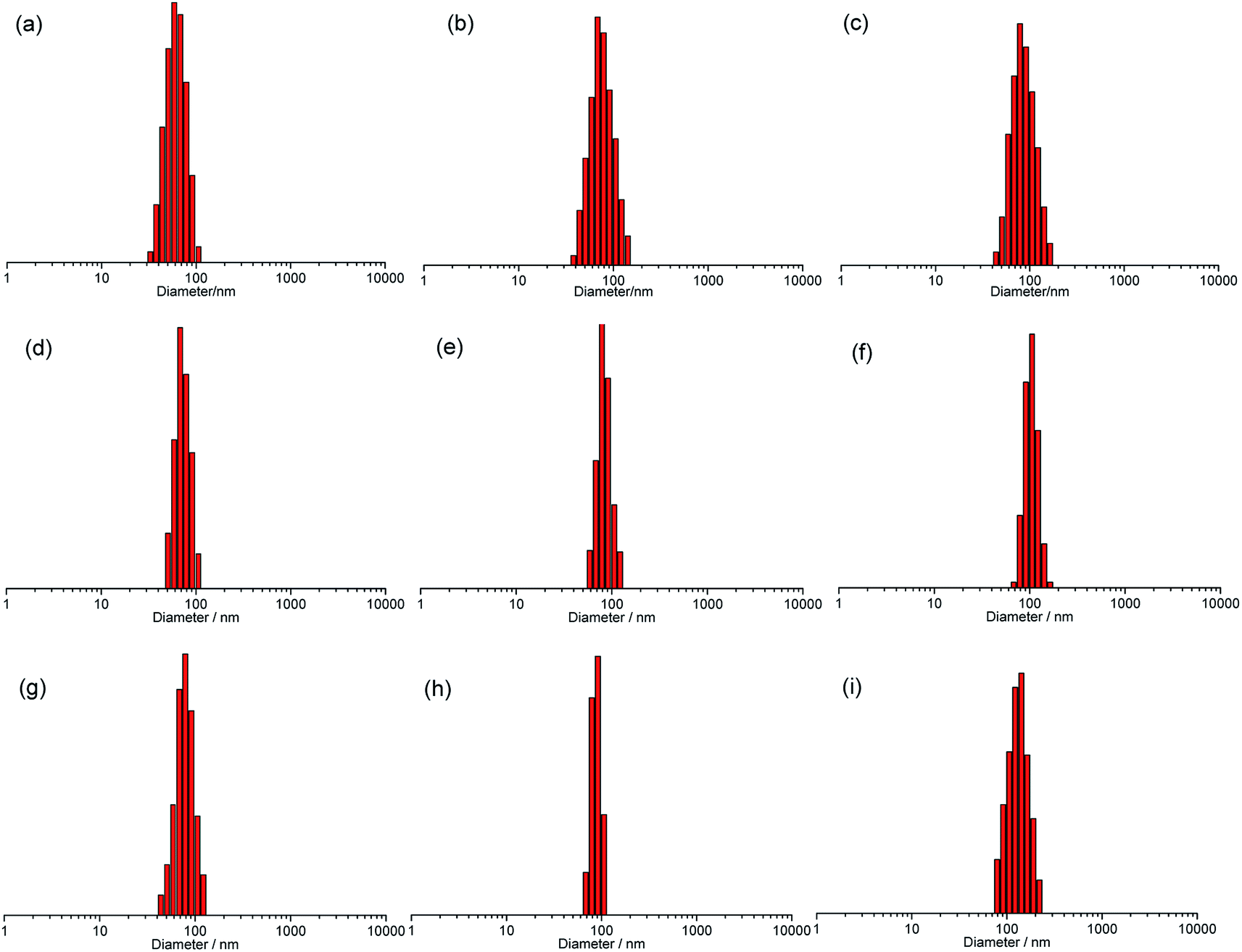
Further insight into the self-assembled morphology of the copolymer film formed by the solvent evaporation in air was carried out through the TEM analysis. TEM image showed that the non-ionic P225 has no self-assembled nanostructures (Fig. S10, ESI†), which was different from that of P125.34 In Fig. S11 (ESI†), the ionic P325 can self-assemble into the solid nanospheres with the diameter of about 20 nm and an uniform distribution at 0.005 mg mL−1 in THF. As shown in Fig. 5 and S12 (ESI†), the non-ionic copolymer PFNP50-b-P225-b-PFNP50 had the self-assembled solid nanospheres with various diameter of about 40, 90, and 110 nm (Fig. 5a–c) at the concentrations of 0.005, 0.01, and 0.02 mg mL−1, respectively.
 | ||
| Fig. 5 TEM images of PFNP50-b-P225-b-PFNP50 (a–c) and PFNP50-b-P325-b-PFNP50 (d–f) at 0.005 (a and d), 0.01 (b and e), and 0.02 (c and f) mg mL−1 in THF; (g) the schematic process of a tree ring-like nanostructure by the self-assembly of triblock copolymer in THF. | ||
Interestingly, the TEM image of the ionic copolymer PFNP50-b-P325-b-PFNP50 showed a tree ring-like nanostructure consisted of alternating P3 and PFNP blocks. This was indicative of the intriguing self-assembly process driven by: the π–π stacking between the aromatic PBI linkers and the interionic attraction made ladderphane P3 blocks closer to each other; meanwhile, the ladderphane P3 blocks were wrapped by the PFNP blocks and away from THF, and formed the small assemblies for the reason that the solubility diversity of the two different blocks in PFNP50-b-P325-b-PFNP50; afterwards, a large number of small assemblies interacted with each other to build up hollow spheres with the regular alternative nano rings to lower the surface energy; at last, some hollow spheres combined with the others so as to eventually achieve a tree ring-like nanostructure with different diameters by the interaction of π–π stacking between the aromatic side groups of the PFNP blocks. The electron cloud density of ladderphane P3 block was higher than the PFNP block because of the larger aromatic conjugated structure of ladderphane P3, resulting in the observed morphology with alternative black region belonging to ladderphane P3 blocks and gray region belonging to PFNP blocks. The thickness of black region was slightly larger than the gray region. The morphology forming process was schematically illustrated in Fig. 5g. For PFNP50-b-P325-b-PFNP50, the diameter of the tree ring-like nanostructure is nearly 50 nm at 0.005 mg mL−1 (Fig. 5d), and the ring number of about 7. As the concentration increased to 0.01 mg mL−1, the tree ring-like nanostructure grew up into larger one with a diameter of about 60 nm with the ring number of about 10 (Fig. 5e). When the concentration was further up to 0.02 mg mL−1, the diameter of the nanostructure increased to about 70 nm with the ring number of about 11 (Fig. 5f). The thicknesses of black region and gray region were about 4 and 3 nm, respectively. In conclusion, the diameter and the ring number of the tree ring-like nanostructure gradually increased with the concentration increase. Importantly, the unique microstructure was conducive to promote the dielectric properties of polymers. To further decrease the dielectric loss, the ionic copolymers PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 with more longer insulating PFNP blocks were prepared. From the TEM images (Fig. S13 and S14, ESI†), the diameters of the tree ring-like nanostructure for PFNP100-b-P325-b-PFNP100 were 53, 57, and 70 nm at 0.005, 0,01 and 0.02 mg mL−1, respectively, which was closed to those of PFNP50-b-P325-b-PFNP50. It may be because that when the nanostructures existed in THF, flexible PFNP blocks was loosely packed around the rigid P3 blocks due to the good solubility in THF so that the copolymer with the longer PFNP blocks can form the nanostructures with larger Dhs, but as the solvent evaporation, the flexible PFNP blocks were easier to collapse than the rigid P3 blocks, and the length of PFNP blocks was no longer a decision factor to influence the size of the nanostructures relative to the P3 blocks. Therefore, the size of PFNP100-b-P325-b-PFNP100 was similar to that of PFNP50-b-P325-b-PFNP50 because the ionic copolymers possessed the same length of rigid P3 blocks, while the alternately dark and bright nanostructure of PFNP100-b-P325-b-PFNP100 became less visible. The ring numbers of nanostructure from PFNP100-b-P325-b-PFNP100 were about 7, 8, and 9 at 0.005, 0,01 and 0.02 mg mL−1, respectively. The thicknesses of black region and gray region were nearly 5 and 3 nm, respectively. The nanostructure of PFNP200-b-P325-b-PFNP200, which was different from the above two, was mostly irregular shape, and only a small amount of the tree ring-like nanostructure could be found at 0.02 mg mL−1, likely due to its self-assembly ability weakened as increasing the length of PFNP blocks and the solubility of copolymer. The size of the nanostructures estimated by DLS were larger than that by TEM, which was in agreement with that the size of the dried sample was somewhat smaller than that in solution.
To determine whether or not the block copolymers could self-assemble into a tree ring-like nanostructure in THF solution, a frozen-treatment for polymer solution was adopted: a copper grid coated with carbon was put into a tube with full of nitrogen, the solution of ionic copolymer PFNP50-b-P325-b-PFNP50 in THF at a concentration of 0.005 mg mL−1 was dropped onto the copper grid, and then the tube was put into the liquid nitrogen for 15 minutes, which made the solution into a frozen state to prevent THF from volatilizing; at last, THF was removed under reduced pressure to afford a solid polymer sample for TEM testing. The TEM image showed still a tree ring-like morphology with a diameter of about 55 nm (Fig. S15, ESI†), which proved the block copolymers can self-assemble into a tree ring-like nanostructure in THF solution.
Dielectric properties of ladderphanes
The dielectric performance of polymers was closely related to their polar group and the chain microstructure. Fig. 6 displayed the dielectric spectroscopy of the polymers with a thin film thickness of about 1–80 μm. For homopolymers P125 and P225 with similar molecular weight, at a frequency range of 100 Hz to 1 MHz, the observed different values of dielectric response were 5 and 7, respectively. One reason could explain this phenomenon that the rigid pyrrolidine was aligned coherently in one orientation during the ROMP process, which was beneficial for the positive accumulation of dipole moments during the orientation of polar pendants under an electric field.10 The dielectric loss for these two polymers was in the range of 0.02–0.04. By introducing PFNP block, the dipole moment of polymer increased due to introducing polar F group; meanwhile, the solubility of copolymer was improved to facilitate the preparation of thin film layer free from a lot of defects. Copolymer PFNP50-b-P225-b-PFNP50 had a relatively higher dielectric constant of 12 and a lower dielectric loss of 0.02–0.03 in comparison to P225. The dielectric loss mostly came from the motion of chain in electric field below glass transition temperature11 and the defects of polymer film.47 | ||
| Fig. 6 Dielectric constant (a) and dielectric loss (b) of polymers versus frequency from 100 Hz to 1 MHz. | ||
The polarization of ionic P325, as shown in Scheme S3,† which was from small relative displacements of PF6− under the applied electric field, presented a much higher dielectric constant value ranging from 22 to 24 than the non-ionic polymers below 1000 Hz, because the induced polarization from the surrounding ions and the orientation polarization of the ion could increase the molecular dipoles effectively.38 With the increase of frequency, the dielectric constant of ionic P325 decreased to 18 because the induced polarization and orientation polarization faded away, and the value was still higher than that of the corresponding non-ionic P225 likely due to the ionic group with a high dipole moments. The dielectric loss of P325 was in the range of 0.09–0.13 below 1000 Hz because the orientation displacements of ionic groups lagged the frequency of alternating electric field20 and there was a serious leakage current.21 When the frequency of the applied electric field was over 1000 Hz, due to the dielectric loss was positively correlated with the conductivity,48 the dark current caused an increased dielectric loss value from 0.09 at 1000 Hz to 0.12 at 1 MHz by the ionic conduction.21 Therefore, it was a practical way to decrease the dielectric loss of ionic polymers by inhibiting ionic conduction. The ionic copolymer PFNP50-b-P325-b-PFNP50 with a tree ring-like nanostructure had a high dielectric constant value ranging from 23 to 21 accompanied with the dielectric loss decrease from 0.08 to 0.06 as the frequency varied from 100 Hz to 1000 Hz. When the frequency further increased to 1 MHz, the dielectric constant of ionic copolymer still be held at 18, while the dielectric loss reduced to 0.06–0.07 and was much lower than those of ionic P325, which indicated that the dark current was restrained effectively. With the increase of insulating PFNP block ratios, the dielectric constant and dielectric loss of PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 were reduced to 18 and 17, as well as 0.05–0.04 and 0.04–0.03 at a frequency range of 100 Hz to 1 MHz, respectively, but they were stable and almost independent of the frequency between 100 Hz and 1 MHz, which had a better practical application in comparison to P3 and PFNP50-b-P325-b-PFNP50. When the length of the insulating PFNP blocks increased beyond a certain point, it was more effective to make the ionic ladderphane P3 blocks isolated and inhibit the transport of ions to reduce the current leakage under an applied electric field.12 Besides, the solubility of ionic copolymer was improved to facilitate the preparation of polymer thin film layer free from a lot of defects by increasing the PFNP block length (Fig. S16, ESI†), resulting in improved dielectric properties.
The maximum field strength and maximum polarization of P225 and PFNP50-b-P225-b-PFNP50 were 570 and 590 MV m−1, as well as 0.31 and 0.39 μc cm−2, respectively, as shown in Fig. 7a and b. The linear polarization behavior and low hysteresis indicated the low level of energy dissipation, which was in good agreement with the low dielectric loss in the impedance test. However, the low polarization is a disadvantage in thin film capacitors. In contrast, the maximum field strength of ionic P325, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200 as shown in Fig. 7c–f were down to 75, 160, 214, and 207 MV m−1, but the maximum polarization increased to 0.35, 0.96, 1.28, and 1.24 μc cm−2 sequentially in comparison to the non-ionic polymers at the same field strength due to the ionic polarization. Although having a high polarization, the hysteresis loop with dipole saturation of the ionic homopolymer P325 was large at high fields due to the serious leakage current,47 and the ion migration was responsible for the hysteresis. For ionic copolymer PFNP50-b-P325-b-PFNP50, it can self-assemble into the tree ring-like nanostructure, where the ionic P3 blocks were isolated between the insulating PFNP blocks and the long distance migration of ion is difficult to achieve. Importantly, the hysteresis loops of ionic copolymers PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200 were effectively narrowed by increasing the insulating PFNP blocks, and the current leakage and the energy loss were reduced.
 | ||
| Fig. 7 Polarization (D–E) loops for P225 (a), PFNP50-b-P225-b-PFNP50 (b), P325 (c), PFNP50-b-P325-b-PFNP50 (d), PFNP100-b-P325-b-PFNP100 (e), and PFNP200-b-P325-b-PFNP200 (f). | ||
The energy density (Ue) of a capacitor was given by the equation:12  , where E and D were the applied electric field and electric displacement, respectively. According to this equation, the stored energy density (Us) or released energy density (Ur) of the polymers as a function of the maximum electric field strength could be obtained by integrating the charge or discharge curve of the D–E loop. The charge–discharge efficiency (η) was given by the equation: η = (Ur/Us) × 100%. As shown in Fig. 8a and b, the Us and Ur of non-ionic P225 and PFNP50-b-P225-b-PFNP50 were 0.95 and 0.89 J cm−3, as well as 1.16 and 1.12 J cm−3 under 570 and 590 MV m−1, respectively. The η values of non-ionic polymers were larger than 94% by the calculation. Ionic polarization effectively increased the energy density of the polymers. For ionic P325, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200, the Us and Ur values as shown in Fig. 8c–f were changed to 0.21, 0.96, 1.50, and 1.35 J cm−3 as well as 0.03, 0.67, 1.31, and 1.21 J cm−3 under 75, 160, 214, and 207 MV m−1, respectively.
, where E and D were the applied electric field and electric displacement, respectively. According to this equation, the stored energy density (Us) or released energy density (Ur) of the polymers as a function of the maximum electric field strength could be obtained by integrating the charge or discharge curve of the D–E loop. The charge–discharge efficiency (η) was given by the equation: η = (Ur/Us) × 100%. As shown in Fig. 8a and b, the Us and Ur of non-ionic P225 and PFNP50-b-P225-b-PFNP50 were 0.95 and 0.89 J cm−3, as well as 1.16 and 1.12 J cm−3 under 570 and 590 MV m−1, respectively. The η values of non-ionic polymers were larger than 94% by the calculation. Ionic polarization effectively increased the energy density of the polymers. For ionic P325, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200, the Us and Ur values as shown in Fig. 8c–f were changed to 0.21, 0.96, 1.50, and 1.35 J cm−3 as well as 0.03, 0.67, 1.31, and 1.21 J cm−3 under 75, 160, 214, and 207 MV m−1, respectively.
 | ||
| Fig. 8 Energy density versus applied electric field for P225 (a), PFNP50-b-P225-b-PFNP50 (b), P325 (c), PFNP50-b-P325-b-PFNP50 (d), PFNP100-b-P325-b-PFNP100 (e), and PFNP200-b-P325-b-PFNP200 (f). | ||
Evidently, ionic polymers possessed a higher energy density than the non-ionic counterparts. Besides, the Us values of ionic copolymers were much higher than that of a commercial BOPP capacitor film (less than 0.50 μC cm−2) at 200 MV m−1.12 However, the ionic homopolymer P325 was accompanied by an increase in energy loss due to the serious current leakage inside the conductive segments, and this resulted in so low Ur of 0.03 J cm−3. The η of P325 was lower than 15% under 75 MV m−1, and the dissipative energy during the charge and discharge cycles is a disadvantage in applications.21 Compared to P325, the ionic copolymers PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP20 had the higher η of 69.8, 87.3, and 89.6% under 160, 214, and 207 MV m−1, respectively. Especially for PFNP100-b-P325-b-PFNP100 and PFNP200-b-P325-b-PFNP200, the ionic P3 blocks were effectively inhibited by the insulating PFNP blocks so that it was hard for them to associate with each other, and they should be better candidates for polymer dielectric materials with a high energy density and low dissipation at room temperature.
Conclusions
In summary, the PBI-bridged homopolymer ladderphanes, P125, P225, and P325, as well as the triblock copolymer ladderphanes, PFNP50-b-P225-b-PFNP50, PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200 were synthesized by ROMP, and their dielectric constants were 5, 7, 21, 12, 20, 18, and 17 accompanied with the dielectric losses of 0.02–0.04, 0.02–0.04, 0.09–0.13, 0.02–0.03, 0.06–0.07, 0.04–0.05, and 0.03–0.04 at a frequency range of 100 Hz to 1 MHz, respectively. The ionic groups in P3 with a high dipole moment could effectively increase the dielectric constant and energy density of polymers, but it also led to a high dielectric loss due to the serious leakage current. The multi-ringed nanostructure provided a beneficial effect that the ionic blocks were isolated between the insulating blocks, and the long distance migration of ion was difficult to achieve, which effectively inhibited the current leakage and reduced the hysteresis loop. Thus, the ionic block copolymers PFNP50-b-P325-b-PFNP50, PFNP100-b-P325-b-PFNP100, and PFNP200-b-P325-b-PFNP200 with the self-assembled tree ring-like nanostructures could achieve a high dielectric constant and keep a low dielectric loss. This research presented a practical way to improve the dielectric properties by combining the ionic, dipolar, and nano-interfacial polarizations as well as the stereoregular chain microstructure. It was expected to be a good potential dielectric material in the use of thin film capacitor.Acknowledgements
The research was financially supported by the National Natural Science Foundation of China (No. 21574041, 21374030, 21074036) and the Large Instruments Open Foundation of East China Normal University (No. 20151006).Notes and references
- M. Guo, X. Yan, Y. Kwon, T. Hayakawa, M. A. Kakimoto and T. Goodson, J. Am. Chem. Soc., 2006, 128, 14820–14821 CrossRef CAS PubMed.
- M. Guo, T. Hayakawa, M. A. Kakimoto and T. Goodson, J. Phys. Chem. B, 2011, 115, 13419–13432 CrossRef CAS PubMed.
- M. S. Islam, Y. Qiao, C. Tang and H. J. Ploehn, ACS Appl. Mater. Interfaces, 2015, 7, 1967–1977 CAS.
- U. H. Choi, A. Mittal, T. L. Price, H. W. Gibson, J. Runt and R. H. Colby, Macromolecules, 2013, 46, 1175–1186 CrossRef CAS.
- V. K. Thakur, E. J. Tan, M. F. Lin and P. S. Lee, Polym. Chem., 2011, 2, 2000–2009 RSC.
- Q. Wang and L. Zhu, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1421–1429 CrossRef CAS.
- M. Groenewolt, T. Brezesinski, H. Schlaad, M. Antonietti, P. W. Groh and B. Ivan, Adv. Mater., 2005, 17, 1158–1162 CrossRef CAS.
- W. Hao, J. Zhang, Y. Tan, M. Zhao and C. Wang, J. Am. Chem. Soc., 2011, 94, 1067–1072 CAS.
- Z. You, D. Gao, O. Jin, X. He and M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 1292–1301 CrossRef CAS.
- Z. You, W. Song, S. Zhang, O. Jin and M. Xie, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 4786–4798 CrossRef CAS.
- J. Wei, Z. Zhang, J. K. Tseng, I. Treufeld, X. Liu, M. H. Litt and L. Zhu, ACS Appl. Mater. Interfaces, 2015, 7, 5248–5257 CAS.
- W. Liu, X. Liao, Y. Li, Q. Zhao, M. Xie and R. Sun, Chem. Commun., 2015, 51, 15320–15323 RSC.
- A. Kahouli, O. Lebedev, M. Ben Elbahri, B. Mercey, W. Prellier, S. Riedel, M. Czernohorsky, F. Lallemand, C. Bunel and U. Luders, ACS Appl. Mater. Interfaces, 2015, 7, 25679–25684 CAS.
- K. Yang, X. Huang, J. He and P. Jiang, Adv. Mater. Interfaces, 2015, 2, 1500361–1500371 CrossRef.
- X. Huang and P. Jiang, Adv. Mater., 2015, 27, 546–554 CrossRef CAS PubMed.
- M. Molberg, D. Crespy, P. Rupper, F. Nüesch, J. A. E. Månson, C. Löwe and D. M. Opris, Adv. Funct. Mater., 2010, 20, 3280–3291 CrossRef CAS.
- Y. Qiao, M. S. Islam, K. Han, E. Leonhardt, J. Zhang, Q. Wang, H. J. Ploehn and C. Tang, Adv. Funct. Mater., 2013, 23, 5638–5646 CrossRef CAS.
- M. N. Almadhoun, M. N. Hedhili, I. N. Odeh, P. Xavier, U. S. Bhansali and H. N. Alshareef, Chem. Mater., 2014, 26, 2856–2861 CrossRef CAS.
- J. K. Yuan, S. H. Yao, Z. M. Dang, A. Sylvestre, M. Genestoux and J. Bai, J. Phys. Chem. C, 2011, 115, 5515–5521 CAS.
- J. Tang, M. Radosz and Y. Shen, Macromolecules, 2008, 41, 493–496 CrossRef CAS.
- L. Zhu, J. Phys. Chem. Lett., 2014, 5, 3677–3687 CrossRef CAS PubMed.
- H. Y. Liu, Y. Shen, Y. Song, C. W. Nan, Y. H. Lin and X. P. Yang, Adv. Mater., 2011, 23, 5104–5108 CrossRef CAS PubMed.
- G. Wang, X. Huang and P. Jiang, ACS Appl. Mater. Interfaces, 2015, 7, 18017–18027 CAS.
- B. G. Shin, T. Y. Cho, D. Y. Yoon and B. Liu, Macromol. Res., 2013, 15, 185–190 CrossRef.
- S. Huettner, J. M. Hodgkiss, M. Sommer, R. H. Friend, U. Steiner and M. Thelakkat, J. Phys. Chem. B, 2012, 116, 10070–10078 CrossRef CAS PubMed.
- M. D. Damaceanu, R. D. Rusu, V. E. Musteata and M. Bruma, Polym. Int., 2012, 61, 1582–1591 CrossRef CAS.
- C. Li and H. Wonneberger, Adv. Mater., 2012, 24, 613–636 CrossRef CAS PubMed.
- Z. Lu, B. Jiang, X. Zhang, A. Tang, L. Chen, C. Zhan and J. Yao, Chem. Mater., 2014, 26, 2907–2914 CrossRef CAS.
- C. Li, V. Stepanenko, M. J. Lin, W. Hong, F. Würthner and T. Wandlowski, Phys. Status Solidi B, 2013, 250, 2458–2467 CrossRef CAS.
- V. Percec, M. Peterca, T. Tadjiev, X. Zeng, G. Ungar, P. Leowanawat, E. Aqad, M. R. Imam, B. M. Rosen, U. Akbey, R. Graf, S. Sekharan, D. Sebastiani, H. W. Spiess, P. A. Heiney and S. D. Hudson, J. Am. Chem. Soc., 2011, 133, 12197–12219 CrossRef CAS PubMed.
- W. Yue, A. Lv, J. Gao, W. Jiang, L. Hao, C. Li, Y. Li, L. E. Polander, S. Barlow, W. Hu, S. Di Motta, F. Negri, S. R. Marder and Z. Wang, J. Am. Chem. Soc., 2012, 134, 5770–5773 CrossRef CAS PubMed.
- A. Lv, S. R. Puniredd, J. Zhang, Z. Li, H. Zhu, W. Jiang, H. Dong, Y. He, L. Jiang, Y. Li, W. Pisula, Q. Meng, W. Hu and Z. Wang, Adv. Mater., 2012, 24, 2626–2630 CrossRef CAS PubMed.
- M. Teraguchi, D. Tanioka, T. Kaneko and T. Aoki, ACS Macro Lett., 2012, 1, 1258–1261 CrossRef CAS.
- W. Song, H. Han, J. Wu and M. Xie, Chem. Commun., 2014, 50, 12899–12902 RSC.
- H. C. Yang, S. Y. Lin, H. C. Yang, C. L. Lin, L. Tsai, S. L. Huang, I. W. P. Chen, C. H. Chen, B. Y. Jin and T. Y. Luh, Angew. Chem., Int. Ed., 2006, 45, 726–730 CrossRef CAS PubMed.
- M. M. Huang and H. Weingartner, ChemPhysChem, 2008, 9, 2172–2175 CrossRef CAS PubMed.
- T. W. Smith, M. Zhao, F. Yang, D. Smith and P. Cebe, Macromolecules, 2013, 46, 1133–1143 CrossRef CAS.
- E. I. Izgorodina, M. Forsyth and D. R. Macfarlane, Phys. Chem. Chem. Phys., 2009, 11, 2452–2458 RSC.
- C. M. Chou, S. L. Lee, C. H. Chen, A. T. Biju, H. W. Wang, Y. L. Wu, G. F. Zhang, K. W. Yang, T. S. Lim, M. J. Huang, P. Y. Tsai, K. C. Lin, S. L. Huang, C. H. Chen and T. Y. Luh, J. Am. Chem. Soc., 2009, 131, 12579–12585 CrossRef CAS PubMed.
- J. Zhang, D. Zhu and M. Matsuo, Polymer, 2008, 49, 5424–5430 CrossRef CAS.
- J. A. Love, J. P. Morgan, T. M. Trnka and R. H. Grubbs, Angew. Chem., Int. Ed., 2002, 41, 4035–4037 CrossRef CAS.
- L. Delaude, A. Demonceau and A. F. Noels, Macromolecules, 2003, 36, 1446–1456 CrossRef CAS.
- T. Y. Luh, Acc. Chem. Res., 2013, 46, 378–389 CrossRef CAS PubMed.
- S. Kim and K. M. Miller, Polymer, 2012, 53, 5666–5674 CrossRef CAS.
- B. J. Ash, R. W. Siegel and L. S. Schadler, J. Polym. Sci., Part B: Polym. Phys., 2004, 42, 4371–4383 CrossRef CAS.
- H. A. Klok and S. Lecommandoux, Adv. Mater., 2001, 13, 1217–1229 CrossRef CAS.
- M. Mackey, D. E. Schuele, L. Zhu, L. Flandin, M. A. Wolak, J. S. Shirk, A. Hiltner and E. Baer, Macromolecules, 2012, 45, 1954–1962 CrossRef CAS.
- B. Wang, D. Qin, G. Liang, A. Gu, L. Liu and L. Yuan, J. Phys. Chem. C, 2013, 117, 15487–15495 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures, scheme, IR and NMR spectra, GPC traces, DSC and DLS curves, and TEM images. See DOI: 10.1039/c6ra18029a |
| This journal is © The Royal Society of Chemistry 2016 |