
Charge-assisted intermolecular hydrogen bond formed in coamorphous system is important to relieve the pH-dependent solubility behavior of lurasidone hydrochloride†
Abstract
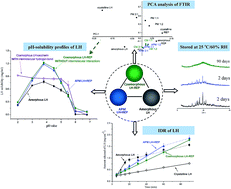
The aim of this study was to enhance the dissolution properties of two BSC II drugs (lurasidone hydrochloride (LH) and repaglinide (REP)) by a coamorphization technique. Coamorphous LH–REP systems (CMs) were prepared by solvent evaporation and characterized by DSC, XRPD, FTIR, Raman spectroscopy, and Ss 13C NMR. The solubilities and intrinsic and supersaturated dissolution profiles, as well as the physical stability, of CMs were compared with the properties of amorphous LH, amorphous REP and their physical mixtures. The single Tg observed in DSC and the disappearance of crystallinity in XRPD indicated the formation of CMs. Principal component analysis of FTIR in combination with Raman spectroscopy and Ss 13C NMR suggested the absence of intermolecular interactions in CMs. In comparison to the pure amorphous forms and their physical mixtures, CMs displayed greatly improved physical stability. In addition, in contrast to the pure amorphous forms, which exhibited temporary enhancements in dissolution, both drugs in CMs exhibited persistent increases in IDRs and supersaturated dissolution, as well as significantly improved solubilities. The persistent pH-dependent solubility behavior of LH in CMs suggested that intermolecular interaction with the N+–H group in the LH structure was probably essential for improving the pH-dependent solubility profile of LH, but was not critical for achieving supersaturated dissolution and preventing the conversion of coamorphous components.
 Please wait while we load your content...
Please wait while we load your content...

