
Alternative role of cisplatin in DNA damage – theoretical studies on the influence of excess electrons on the cisplatin–DNA complex†
Abstract
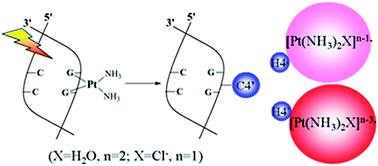
Reliable DFT calculations were used to gain insights into the effects of excess electrons on the cisplatin–DNA complex in a water solution. One electron injection is enough to break the two Pt–N7 bonds, which is driven by the rare symmetrical in-plane bending vibration. The dissociated [Pt(NH3)2]+˙ group from the cisplatin–DNA complex could combine with H2O in the surroundings to form a reactive species, which can abstract the most solvent accessible H4′ of the sugar with a barrier of ca. 17.5 kcal mol−1. Upon influence of the multiple electrons addition, the H4′-abstraction reaction by the stable radical anion is feasible with a lower barrier and is exothermic. Thereby, they have high efficiency for DNA damage. The synergic effect between the metal and the ligand is highlighted due to failure of the isolated [Pt(NH3)2]+˙ and [Pt(NH3)2]−˙ to abstract H4′ of sugar because the overlap between the SOMO (on Pt) and the C4′H4′ anti-bonding orbital is zero. In the present studies, an alternate role of cisplatin in DNA damage was discovered, which strongly confirmed that the cisplatin–DNA complex is more vulnerable to attack from low-energy electrons.
 Please wait while we load your content...
Please wait while we load your content...

