
Guanidiniocarbonyl-pyrrole-aryl conjugates as inhibitors of human dipeptidyl peptidase III: combined experimental and computational study†
Josipa Matića,
Filip Šupljikaa,
Nora Tira,
Patryciusz Piotrowskib,
Carsten Schmuckb,
Marija Abramića,
Ivo Piantanida*a and
Sanja Tomić*a
aDivision of Organic Chemistry and Biochemistry, Ruđer Bošković Institute, 10000 Zagreb, Croatia. E-mail: pianta@irb.hr; sanja.tomic@irb.hr
bInstitute for Organic Chemistry, University of Duisburg-Essen, Universitässtrasse 7, 45141 Essen, Germany. E-mail: carsten.schmuck@uni-due.de; Fax: +49 201 183 4259
First published on 26th August 2016
Abstract
Dipeptidyl peptidase III (DPP III) is a zinc dependant peptidase which catalyses hydrolysis of the second peptide bond from the N-termini of its substrates. DPP III is an enzyme of broad substrate specificity and it has been found in many organisms. It has been recognised in several processes of interest for drug development like pain modulation and defence against oxidative stress. However, its fundamental physiological role is unknown and specific inhibitors would be of significant help in identifying this role. In this work we combined experimental (UV/Vis, fluorimetry and microcalorimetry experiments) with molecular dynamic simulations to study the binding of several newly designed and synthesised guanidiniocarbonyl-pyrrole-aryl conjugates to human DPP III. We found that new compounds bind with micromolar affinity to the enzyme and with varied efficiency inhibit hydrolysis of Arg-Arg-2-naphthylamide, the standard synthetic substrate of DPP III. The molecular modelling study revealed multiple binding modes of the guanidiniocarbonyl-pyrrole-aryl conjugates into the active site of human DPP III. In order to elucidate which one is the most favourable we studied the molecular determinants for their binding to DPP III as well as their influence on protein structure. It seems that the main requirement for a good DPP III inhibitor is a bulky aryl-substituent and a linker of suitable length and flexibility between it and the guanidiniocarbonyl-pyrrole. The obtained results gave directions for future development and improvement of DPP III inhibitors.
Introduction
Dipeptidyl peptidase III (DPP III, EC 3.4.14.4) is a monozinc metalloexopeptidase that hydrolyses dipeptides from the N-termini of its substrates consisting of three or more amino acids. Widely distributed in mammalian tissue, DPP III has been considered to participate in intracellular protein catabolism but it is also indicated in pain modulation1 as well as in the stress response mechanism in mammals.2 DPP III contributes to the activation of transcription factor Nrf2, a constituent of the Nrf2-Keap1 signalling pathway, the main defence mechanism against many environmental toxic agents and carcinogens in cells.3Until now several 3D structures of human DPP III (h.DPP III) have been determined, ligand free and in complexes with natural peptides (PDB_codes 3FVY, 5E33, and 3T6B, 3T6J, 5E3A, 5E2Q, 5EHH and 5E3C, respectively). Human DPP III structure consists of two domains, the catalytic one bearing the metal ion and the larger, “satellite” domain with a deep cleft in between. The presence of the interdomain cleft, together with promiscuous substrate specificity,1 suggested that h.DPP III could experience significant internal motions. The exhaustive in silico study of h.DPP III4–10 revealed that it can adopt a number of different forms in solution, wherein the most compact form was determined as the most stable.6,7 The substrate to be hydrolysed binds preferably into a semi-closed conformation in a way to interact with both domains, influences the mutual orientation of domains and shifts the conformational equilibrium towards more compact protein form. In agreement with the experimental data we found that DPP III prefers ligands that adopt β-strand form and binds to the five-stranded β-core of the enzyme in an antiparallel fashion.
We have shown that two dipeptidyl hydroxamic acids, H-Tyr-Phe-NHOH and H-Tyr-Gly-NHOH act as the competitive inhibitors of the human DPP III at pH 8.0 with the Ki value of 0.15 μM and 10.5 μM, respectively.8 Further on, to examine the influence of amino acid side chain substitution in P1 position of dipeptidyl hydroxamates on their inhibitory potency towards DPP III, a series of H-Phe-X-NHOH compounds was synthesized and the inhibition constants were determined for competitive inhibition of the hydrolysis of Arg-Arg-2-naphthylamide (Scheme 1, 1), the preferred, fluorogenic DPP III substrate. Among them, H-Phe-Phe-NHOH was identified as the most potent inhibitor of human DPP III (Ki = 28 nM) at pH 7.4, followed by H-Phe-Leu-NHOH and H-Phe-Gly-NHOH with Ki values of 0.65 μM and 4.6 μM, respectively.9
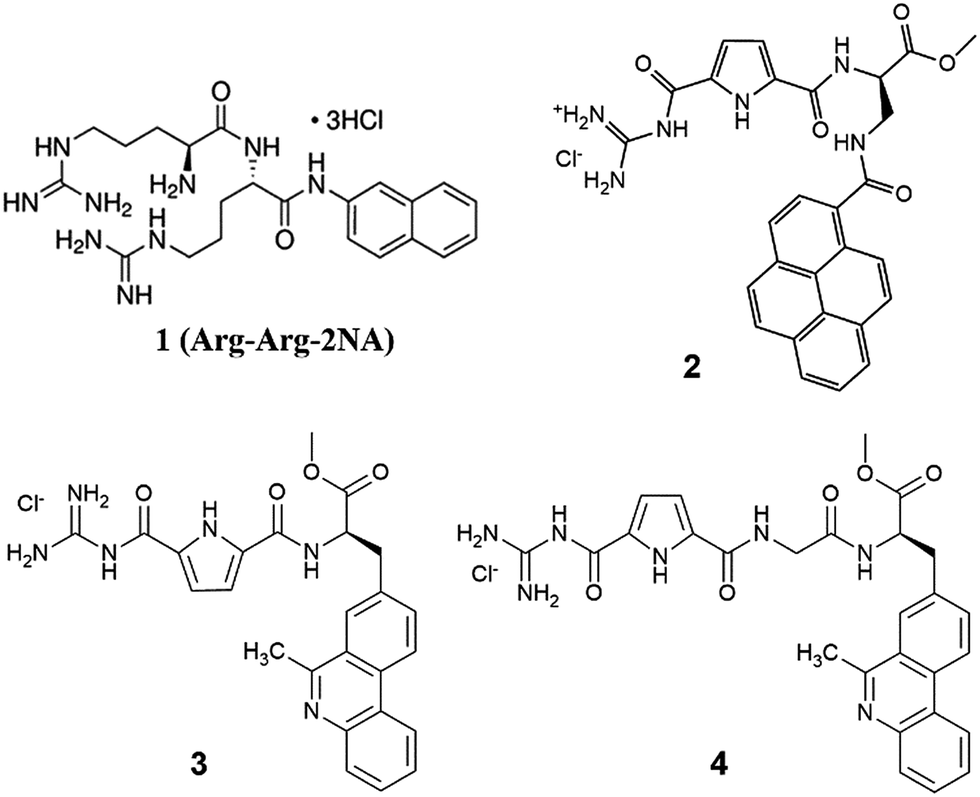 | ||
| Scheme 1 Standard DPP III substrate Arg-Arg-2NA (1) and potential inhibitors: pyrene-guanidiniocarbonyl-pyrrole conjugate (2) and phenanthridine derivatives (3 and 4). | ||
Among many approaches in design of potential enzyme inhibitors, we have chosen as the start point the structure of the standard DPP III substrate, Arg-Arg-2-naphthylamide (Scheme 1, Arg-Arg-2NA). We looked for similar structures (derivatives with guanidine and fluorescent aryl unit), which could bind to the enzyme by similar set of interactions but would not be susceptible to chemical degradation. The general structure of guanidiniocarbonyl-pyrrole moiety11 looked as a good alternative to Arg-Arg-moiety with possible advantage of lower pKa value. Namely, guanidiniocarbonyl-pyrrole moiety with pKa ≈ 6 is neutral at physiological conditions (pH 7.4) and could form strong H-bonds within the enzyme binding site, as well as strong hydrophobic interactions. Moreover some guanidiniocarbonyl-pyrrole analogues already showed promising enzyme inhibition properties.12 Also, within the last years we studied series of guanidiniocarbonyl-pyrrole-aryl conjugates as DNA/RNA targeting molecules,13 whereby recently published pyrene derivative14 (Scheme 1, compound 2) demonstrated to be good fluorimetric reporter to small changes in DNA/RNA binding site structure. Therefore, we decided to explore application of 2, as well as two newly synthesized guanidiniocarbonyl-pyrrole-aryl conjugates based on phenanthridine fluorophore (Scheme 1, compounds 3 and 4), as h.DPP III inhibitors.
In this work we combined experimental, kinetic and microcalorimetry experiments with molecular dynamic simulations to study binding of several guanidiniocarbonyl-pyrrole-aryl conjugates to human DPP III. The study showed that all studied compounds inhibit hydrolysis of Arg-Arg-2NA but with large span of efficiencies, wherein according to the molecular modelling results, they can adopt several different orientations in the h.DPP III active site. The binding free energies, both calculated and measured, revealed exergonic binding of the studied guanidiniocarbonyl-pyrrole-aryl conjugates, similar to binding of the preferred substrate (Arg-Arg-2NA).
Results and discussion
Synthetic procedures
Pyrene derivative 2 was prepared as described earlier.14 Novel phenanthridine derivatives 3 and 4 were prepared starting from previously described Phen-AA15 and Phen-Gly15 by simple peptide coupling procedures shown on Scheme 2. | ||
Scheme 2 Starting from Phen-AA15 and Phen-Gly15 by reaction procedure (i) HBTU, HOBT, Et3N, CH3CN, r.t.; (ii) TFA![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :H2O/9:1, CH2Cl2, r.t.; were prepared 3 and 4. :H2O/9:1, CH2Cl2, r.t.; were prepared 3 and 4. | ||
Study of interactions of novel compounds with h.DPP III
Aqueous solution of compound 2 was previously characterised.14 Novel analogues 3 and 4 showed to be stable in aqueous solutions and the absorbency of buffered solution of 3 and 4 was proportional to their concentration within the used concentration range, pointing out that no aggregation took place at experimental conditions used. Aqueous solutions of 2–4 emit intrinsic fluorescence, intensity proportional to the compound concentration up to 10 micromolar concentrations.In order to determine the binding affinities of novel compounds, in comparison to the affinity of the substrate Arg-Arg-2NA, to h.DPP III titration experiments were performed with its inactive E451 mutant.
We have chosen two complementary titration techniques (microcalorimetry and fluorescence), while keeping the same titration procedure: successive additions of compound solution aliquots to protein solution in ITC cell or fluorimetric cuvette. ITC was used to monitor the change of heat as a result of the compound–protein complex formation, while for fluorophores (2, 3 and 4) we monitored the changes in compound fluorescence, particularly the emission spectrum shape and possible non-linearity of emission intensity in dependence on the compound concentration.
For instance, the fluorescence spectrum of 2, particularly the relative ration between emission maxima at 387 and 404 nm, significantly changed in the presence of enzyme (Fig. 1). All titration data were processed for stoichiometry [compd]/[h.DPP III mutant E451A] = 1 (in accordance with the literature data for referent Arg-Arg-2NA) to give binding constants (Table 1). Under such conditions ITC results revealed almost identical binding affinity values as did the fluorimetric titration experiment (Fig. 1, Table 1). The obtained results showed excellent agreement between logKa values obtained with two independent methods, and also revealed that all studied compounds show comparable, and biologically relevant, affinity toward the studied protein.
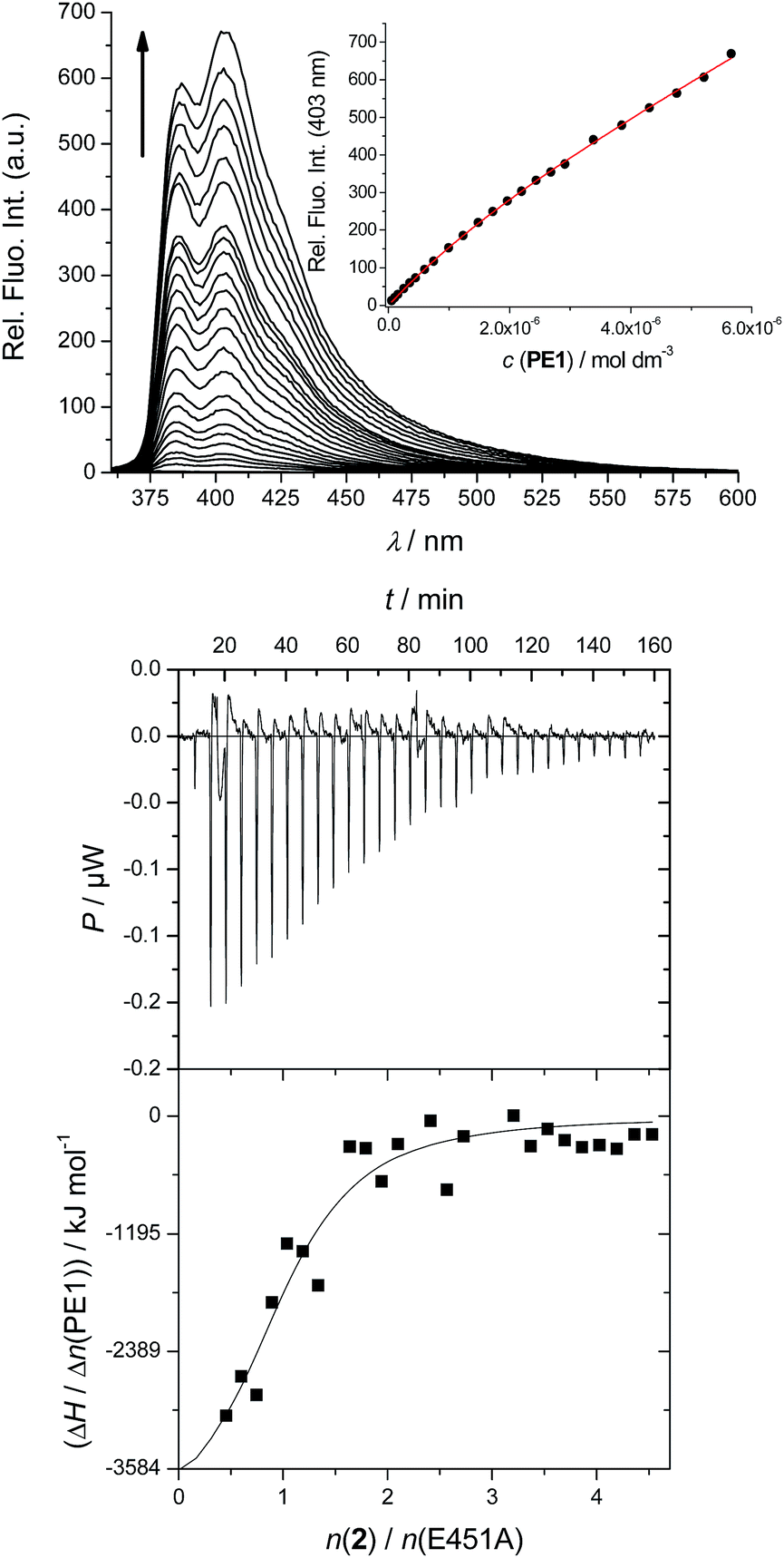 | ||
| Fig. 1 Titration of the h.DPP III mutant E451A with 2: UP: fluorimetric titration (c(enzyme) = 1 × 10−6 M, λexc = 342 nm); DOWN: ITC titration (c(enzyme) = 2.5 × 10−6 M). Done at pH = 7.4, 20 mM Tris-HCl buffer. | ||
Ka)a calculated from the ITC and fluorimetric titrations of h.DPP III mutant E451A with compounds 2, 3 and 4, and ITC titration of referent Arg-Arg-2NA. Measurements were done in 20 mM Tris-HCl buffer pH = 7.4
| logKa (fluo) |
logKa (ITC) |
ΔrH/kJ mol−1 | ΔrS/J K−1 mol−1 | ΔrG/kJ mol−1 | |
|---|---|---|---|---|---|
| a The titration data were fitted for stoichiometry [compd]/[h.DPP III mutant E451A] = 1 by Origin (ITC) and Specfit16 (complete spectra of fluorimetric titrations). | |||||
| 2 | 5.8 | 6.0 | −18.1 | 54.1 | −34.2 |
| Arg-Arg-2NA | — | 5.9 | −23.1 | 34.6 | −33.4 |
| 3 | 5.8 | 5.6 | 30.6 | 210.0 | −32.0 |
| 4 | 5.5 | — | |||
Detailed analysis of ITC data indicated different thermodynamics of binding of the inhibitors 2 and 3 to the enzyme. While binding of 2 to h.DPP III is characterised by negative ΔrH, similarly to the binding of Arg-Arg-2NA to the enzyme, the binding of phenanthridine derivative 3, is characterised by positive ΔrH. Intriguing difference in enthalpic response between close analogues 2 and 3 is most likely related to different size of aryl moiety, and also possible influence might come from the heterocyclic nitrogen of 3. However, reason for the significant increase of entropy upon binding of 3 to DPP III, in comparison with that of 2, is not clear.
Inhibition studies with h.DPP III wt
During the timespan typical for the Arg-Arg-2NA hydrolysis catalysed by h.DPP III (15 min on 37 °C) none of the studied compounds showed measurable change in ITC measurements. Moreover, the fluorescence emission of 2–4 didn't change during 15 min of preincubation of compounds in buffer (50 mM Tris-HCl, pH = 7.4) with the enzyme. Apparently the studied compounds are not hydrolysed by enzyme, although their binding affinities are comparable with that of Arg-Arg-2NA (Table 1).Examination of the impact of 2–4 on the activity of human DPP III was performed by 15 min of preincubation of the compounds in buffer (50 mM Tris-HCl, pH = 7.4) with the enzyme at 25 °C, followed by activity test with 40 μM Arg-Arg-2NA, 15 min at 37 °C, whereby the extent of Arg-Arg-2NA hydrolysis was measured by colorimetric method5 monitoring the release of 2NA. As a reference was used the same h.DPPIII wt-Arg-Arg-2NA system, but without inhibitors. Analysis of the results (Table 2) showed that all studied compounds significantly inhibited enzymatic hydrolysis of Arg-Arg-2NA, whereby 2 was the most active inhibitor with inhibitory potency comparable to those of previously studied dipeptidyl hydroxamic acids inhibitors, H-Phe-Leu-NHOH and H-Phe-Gly-NHOH.10 Furthermore, significant differences in inhibition efficiency, with the tendency 2 > 3 > 4 (Table 2), do not match rather similar binding constants (Table 1) of the studied compounds. Apparently, the inhibition mechanism besides the strength of non-covalent binding involves some other contributions as well. This presumption was also supported by different thermodynamic signature of binding (Table 1), whereby effects of 2 and Arg-Arg-2NA were very similar at variance to opposite enthalpic effect of phenanthridine analogue (3).
| Compound | c(compound)/μM | % of inhibition |
|---|---|---|
| 2 | 20 | 80 |
| 10 | 36 | |
| 5 | 20 | |
| 3 | 35 | 45 |
| 4 | 60 | 33 |
Therefore, to shed more light on the possible binding interactions and the structure/activity relations two of the compounds with distinctively different binding parameters (2 vs. 3) were chosen for detailed molecular modelling studies within the enzyme binding site.
Molecular modelling of the complexes between h.DPP III and guanidiniocarbonyl-pyrrole-aryl conjugates
Several complexes between h.DPP III and compounds 2 and 3 were prepared as described in the methodological part. The first set of complexes was obtained by the program AUTODOCK4.2 (Lamarckov genetic algorithm with a box centred at the zinc di-cation) wherein two different forms of h.DPP III were considered: the semi-closed one (the conformation obtained after 72 ns of MD simulations of the open h.DPP III structure, PDB code: 3FVY, at room temperature), and the closed form (PDB code: 3T6B), hereinafter referred as cWTMD and cWT structures, respectively. The four complexes with the highest rank (based on lowest energy and the highest cluster population) were selected for further simulations. Differently from the experimentally determined structure of the complex between the E451A mutant of h.DPP III with tynorphin12 where the aromatic part of the ligand is oriented into the enzyme, AUTODOCK14 predicted binding of the guanidiniocarbonyl-pyrrole-aryl conjugates with the aromatic ring pointing towards the entrance of the deep inter-domain cleft (Fig. S1a–d†).Molecular modelling study performed earlier for DPP III complexes with Arg2NA, Leu-enkephalin and endomorphines 1 and 2 (ref. 8) showed that the substrates preferably bind to the five-stranded β-core of DPP III. Recently, such binding mode was also determined for the opioid peptides (Met and Leu-enkephalin) as well as for angiotensin-II and the peptide inhibitor IVYPW by X-ray crystallography.29
The binding modes predicted by AUTODOCK for compounds 2 and 3 significantly differ from the described one. Because of that, we constructed a several additional complexes for each ligand, 2 and 3, using the crystal structures of the DPP III-peptide complexes with peptides as templates (Fig. S1e and f†).
In order to elucidate influence of ligands on the protein structure, we monitored several geometric parameters: RMSD, radius of gyration (Rg) and distances between D186-S500 and Q400-S500 Cα atoms, d1 and d2, respectively (for definition of distances d1 and d2 see Fig. S2† and methods). During the simulations ligand and enzyme, particularly the amino acid residues from its active site, adapted to each other forming stable complexes. While compactness of the complexes obtained using the semi-closed enzyme structure significantly decreased during the simulations, compactness of the complexes with initially closed enzyme structure slightly increased (Fig. S3†), or has not changed. For example the mutual orientation of two domains remained mostly stayed similar during the simulations of complex cWT-2 (Fig. 2).
 | ||
| Fig. 2 Profile of distances d1 and d2 determined during simulations of the cWT-2 complex. | ||
According to the calculated RMSD values and distance d1 and d2 profiles (Fig. S4†), the large aromatic system (pyrene) at the inter-domain cleft entrance (Fig. S5†) complexes cWTMD-2(AD1) and cWTMD-2(AD2) perturbs the enzyme structure more than when pointing towards the protein interior (Fig. S5†). Besides, in the cWTMD-2(AD1) complex the metal ion coordination was perturbed as well. Interestingly, while during the simulations of the cWTMD-2(AD1) complex inhibitor moved away from the zinc ion and its position in the zinc coordination sphere was replaced by a water molecule, during the simulations of cWT-2′ complex ligand moved towards the zinc ion and after about 10 ns established its coordination with Zn2+ (see Fig. 3 and Fig. 6 S6†).
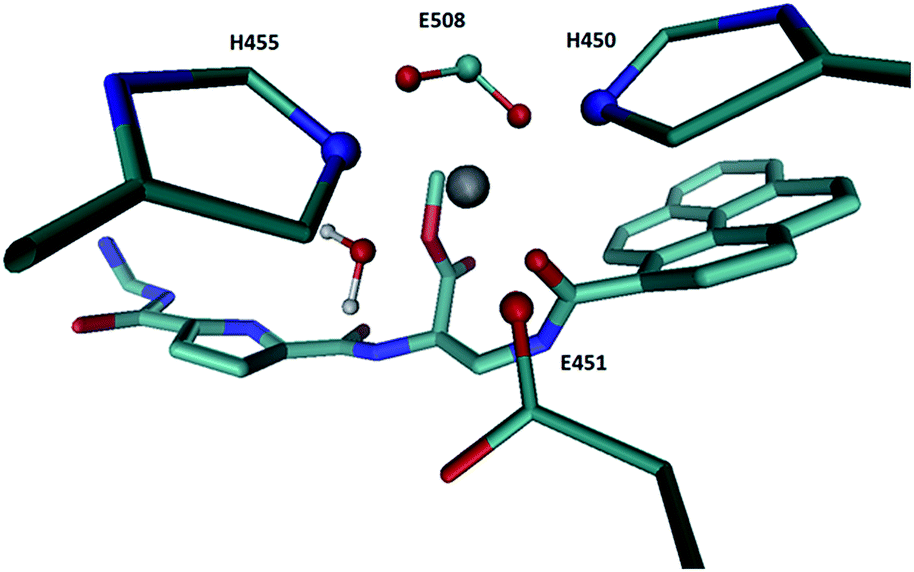 | ||
| Fig. 3 Zn2+ coordination in the structure of the cWT-2′ complex obtained at the end of MD simulations. | ||
In this complex 2 closely resemble the Arg-Arg-2NA position in the h.DPP III active site (see Fig. 4a) which is in the enzyme binding pocket accommodated similarly to the opioid peptides (see Fig. 4b). The pyrene is, similarly to the naphthalene ring of Arg-Arg-2NA accommodated in the S1′ binding subsite, where it is stabilized by Phe443, Val447, His450, as well as amino acid residues from the other subsites: Asp508, His568, Arg669 and Glu316 (Fig S7†). Guanidinocarbonyl-pyrrole is interacting with the residues from S1 and S2 subsite, Tyr196, Glu316, Ser317, Tyr318, Asn394, Asp396, Arg399, Asp496, wherein the guanidine is similarly to the guanidine group of the first, N-terminal arginine of Arg-Arg-2NA stabilized by two strong electrostatic interactions with the carboxyl groups of Asp396 and Asp496. The linker between pyrrole and pyrene is interacting with Ile390, and Asn394 belonging to the β core in the lower DPP III domain as well as with Glu316, Tyr318, and the catalytic glutamates 451 and 508. However, the peptide bond in 2 is oriented differently than in the substrates and so, it is not appropriate for the hydrolysis (see Fig. 4a), i.e. the water molecule hydrogen bonded to Gle451 is too far from the amide carbon to perform the nucleophile attack.
 | ||
| Fig. 4 Structural alignment of TOP: inhibitor 2 (black) and the synthetic substrate Arg-Arg-2NA (cian) bound into the DPP III interdomain cleft. Ligands and Glu451 are given in stick representation, the zinc ion is represented by pink sphere, and the water molecule hydrogen bonded to Glu451 is given in ball and stick representation; BOTTOM: substrates, the synthetic one, Arg-Arg-2NA (cian) and Leu-enkephalin, Tyr-Gly-Gly-Phe-Leu (black) bound into the DPP III interdomain cleft with the amino acid residues Ala388, Gly389, Ile390 and Asn391 belonging to the DPP III β core displayed. The amino acid residues that form the β-strand at bottom of the DPP III active site are represented as thin sticks. | ||
Similarly as during simulations of the h.DPP III–2 complexes, compactness of the h.DPP III–3 complexes with semi-closed enzyme structure significantly decreased during the simulations, while compactness of the complexes with initially closed enzyme structure increased.
However, this ligand perturbed the enzyme structure more severely than 2 did (Fig. S3 and S8†), especially in the complexes where the large aromatic ring is situated at the entrance of the interdomain cleft. Reason for this is its higher rigidity, but also difference in aromatic system (phenanthridine vs. pyrene). In the cWTMD-3 and cWT-3 complexes, position of 3 is similar to the Arg-Arg-2NA position in the enzyme active site. The guanidine group is also stabilized by strong electrostatic interactions with Glu393 and Glu493, phenanthridine is besides by the face to edge interaction with Phe443 stabilized by interactions with Asn406, Leu413, Val447 and Glu451. The phenanthridine to pyrrole linker is like 2 and the earlier studied DPP III substrates stabilized by interactions with the residues from the DPP III β core, as well as with Glu508 and His568 (Fig. S9†). However, like in the case of the cWT-2 complex in none of the h.DPP III–3 complexes there is a water molecule hydrogen bonded to Glu451 that can perform the nucleophile attack to the amide carbon. Absence of the water molecule appropriate for the peptide bond hydrolysis in DPP III complexes with inhibitors is in agreement with the findings of Kumar et al.29
Conclusions
We designed, synthesized and studied three novel structural analogues of standard DPP III substrate (ArgArg-2NA). The novel compounds 2–4 are characterised by replacement of aliphatic guanidines with the more acidic pyrrole guanidine and by introduction of larger aryl moieties (pyrene and phenanthridine). All novel compounds (2–4) bind to h.DPP III with similar affinities as Arg-Arg-2NA, thus showing that designed structural changes do not disturb their binding ability. The most important, novel compounds (2–4) revealed resistance to the DPP III-catalysed degradation in conditions used for Arg-Arg-2NA and moreover it was shown that they inhibit the Arg-Arg-2NA hydrolysis. The results indicate that while increasing the linker length between aryl- and COOMe group boosted the inhibition efficiency of 2 with respect to 3, increasing of the linker length between COOMe group and pyrrole guanidine moiety resulted with decreased inhibition efficiency of 4 with respect to 3 and 2.Detailed study of structure/activity relations showed several intriguing factors: although all studied compounds showed similar binding affinity to DPP III, thermodynamic signature of interactions differed significantly between pyrene (2) and phenanthridine (3) analogue. The molecular modelling study of the inhibitors bound into the enzyme active site cleft also predicted higher stability of the h.DPP III–2 complex in comparison with the complex h.DPP III–3 (phenanthridine derivative). Furthermore, it showed that although the studied guanidiniocarbonyl-pyrrole-aryl conjugates can accommodate into the active site cleft of h.DPP III so that they closely resemble substrate binding, the peptide bond orientation is such that its hydrolysis is not feasible. Interestingly, the water molecules appropriate for the nucleophile attack to the amide carbon has been found in none of the simulated DPP III–guanidiniocarbonyl-pyrrole-aryl conjugate complex. This finding is in line with the previously derived hypothesis (Kumar et al.29) for the peptide inhibitors that their complexes with DPP III lack the water molecule needed for the hydrolysis. Obtained results point that fine tuning of the aryl-guanidinium conjugates structure could further improve efficiency of their interaction with h.DDP III, thus supporting further research within the here presented general structure, which would likely lead to more efficient enzyme inhibitors.
Experimental
General procedures
Reaction solvents and triethylamine were distilled before use. All other reagents were used as obtained from Sigma Aldrich and Alfa Aesar. Thin layer chromatography was performed on Silica Gel 60 F254 plates (Merck; Darmstadt, Germany). Melting points were measured on Kofler hot-stage microscope and are uncorrected. NMR spectra were recorded on Bruker Avance 600 and 300 MHz spectrometers, operating at 150.92 or 75.47 MHz for 13C and 600.13 or 300.13 MHz for 1H nuclei. TMS or DMSO-d6 were used as an internal standard. FT-IR spectra of the samples in KBr pellets were recorded at resolution of 4 cm−1 on an ABB Bomem MB102 single-beam spectrometer and were obtained as KBr pellets. HRMS analysis was performed on MALDI-TOF mass spectrometer operating in reflectron mode. Mass spectra were acquired by accumulating three spectra after 400 laser shots per spectrum. Calibrant and analyte spectra were obtained in positive ion mode. Calibration type was internal with calibrants produced by matrix ionization (monomeric, dimeric and trimeric CHCA), with azithromycin and angiotensin II dissolved in α-cyano-4-hydroxycinnamic acid matrix in the mass range m/z 190.0499 to 749.5157 or 1046.5417. Accurately measured spectra were internally calibrated and elemental analysis was performed on Data Explorer v. 4.9 Software with mass accuracy better than 5 ppm. Samples were prepared by mixing 1 μL of analyte methanol solution with 5 μL of saturated (10 mg mL−1) solution of α-cyano-4-hydroxycinnamic acid (α-CHCA) and internal calibrants (0.1 mg mL−1) dissolved in 50% acetonitrile/0.1% TFA. The UV-Vis spectra were recorded on a Varian Cary 100 Bio spectrometer in quartz cuvettes (1 cm). Fluorescence spectra were recorded on Varian Cary Eclipse fluorimeter in quartz cuvettes (1 cm). Under the experimental conditions used (∼10−6 M) the absorbance and fluorescence intensities of 2–4 were proportional to their concentrations.In fluorimetric experiments, excitation wavelengths at λmax ≥ 305 nm were used in order to avoid absorption of excitation light by protein. Fluorimetric titrations were performed by adding portions of compound solution into the solution of the protein. After mixing protein with studied compounds it was observed in all cases that equilibrium was reached in less than 120 seconds. In following 1 hour fluorescence spectra of complexes remained constant.
Purification of human DPP III
Recombinant human DPP III was obtained by heterologous expression in Escherichia coli and purification according to Špoljarić et al.5DPP III activity assay
The enzymatic activity of wild-type human DPP III was determined by a standard assay at 37 °C with Arg-Arg-2NA as substrate, using the colorimetric method as previously described.5Isothermal titration calorimetry (ITC)
To study the interaction of DPP III with studied compounds by ITC MicroCal VP-ITC microcalorimeter (MicroCal, Inc., Northampton, MA, USA) was performed at pH 7.4, 20 mM tris-HCl buffer. Origin 7.0 software, supplied by the manufacturer was used for data analysis. The reference cell was filled with ultrapure water. Typically, titration was performed in a way that one aliquot of 2 μL and twenty-nine aliquots of 10 μL of the 1–3 (c = 1 × 10−4 M) were injected from rotating syringe (112–270 rpm) into the isothermal cell, equilibrated at 25.0 °C, containing 1421 mL of h.DPP III mutant E451A (c = 5–10 μM). The spacing between each injection was in the range 300–600 s. Initial delay before first injection was in the range 600–3000 s. All solutions used for ITC experiments were degassed prior to use under vacuum (0.64 bar, 10 min) to eliminate air bubbles.Microcalorimetric experiment directly gave three parameters: reaction enthalpy change (ΔrH), binding constant (logKa) and stoichiometry (N). The value of ΔrG was calculated from the binding constant (ΔrG = −RT lnK) and the reaction entropy change was calculated from the binding enthalpy and Gibbs energy (ΔrS = (ΔrH − ΔrG)/T).
Synthesis
Starting material Phen-AA and Phen-Gly (Scheme 2) was prepared as previously described.15:1, v/v; 2 mL) and the reaction was stirred at room temperature overnight. Triflouroacetic salt of deprotected amino acid was obtained as yellow oil after evaporation of the solvent. Deprotected compound was then dissolved in dry acetonitrile (3 mL) and Boc-GCP-OH (35.8 mg, 0.09 mmol), HBTU (34.8 mg, 0.09 mmol, 98%), HOBT (12.5 mg, 0.09 mmol, 97%) and Et3N (75.4 μL, 0.54 mmol) were added. Reaction was stirred at room temperature overnight. Product 3-Boc was isolated by preparative thin layer chromatography in dichloromethane:methanol 9:1 (white solid, 31.8 mg, 63%). To the solution of Boc protected amino acid 3-Boc (17.2 mg, 0.03 mmol) in dichloromethane (4 mL) was added TFA-H2O mixture (9:1, v/v, 2 mL) and the reaction was stirred at room temperature overnight. Solvent was then evaporated and residue purified by preparative thin layer chromatography in dichloromethane:methanol 9:1. Trifluoroacetic salt of deprotected amino acid was obtained as colourless oil, dissolved in methanol (2 mL) and hydrochloric acid (5%, 2 mL) was added. Reaction was stirred at room temperature for 1 hour. Solvent was then evaporated to afford the hydrochloride salt 3 of the deprotected compound as pale yellow solid (14.9 mg, 91%).M.p. > 300 °C (decomposition); IR (KBr) νmax/cm−1: 3400 (s), 3335 (s), 2957 (w), 2920 (w), 2854 (w), 1701 (s), 1645 (m), 1555 (m), 1481 (w), 1442 (w), 1362 (w), 1275 (m), 1227 (w), 1103 (vw), 852 (vw), 814 (vw), 758 (w), 604 (w); 1H NMR (DMSO-d6) δ/ppm: 12.48 (s, 1H, NH–Pyrr), 12.16 (s, 1H, NH–Guan), 9.22 (d, J = 7.7 Hz, 1H, NH–Ala), 9.00–8.85 (m, 2H, CH–Phen), 8.79–8.47 (m, 5H, NH–Guan, NH2–Guan, CH–Phen), 8.37 (bs, 1H, CH–Phen), 8.19 (bs, 1H, CH–Phen), 7.98–7.82 (m, 2H, CH–Phen), 7.56 (s, 1H, CH–Pyrr), 6.89 (s, 1H, CH–Pyrr), 4.97–4.87 (m, 1H, CH–Ala), 3.69 (s, 3H, OCH3), 3.57–3.50 (m, 2H, CH2–Ala), 3.23 (s, 3H, CH3); 13C NMR (DMSO-d6) δ/ppm: 171.61 (COOCH3), 159.52 (Cq), 159.01 (Cq), 155.53 (Cq), 131.62 (Cq), 130.26 (CH–Phen), 128.57 (CH–Phen), 125.86 (Cq), 124.21 (Cq), 123.65 (Cq), 123.37 (CH–Phen), 123.25 (CH–Phen), 115.80 (CH–Pyrr), 113.23 (CH–Pyrr), 53.39 (CH–Ala), 52.15 (OCH3), 36.05 (CH2–Ala); HRMS (MALDI-TOF/TOF): m/z: calcd for C25H24N6O4+: 473.1931, found 473.1941 [M + H]+.
:1, v/v; 1 mL) and the reaction was stirred at room temperature overnight. Triflouroacetic salt of deprotected amino acid was obtained as yellow oil after evaporation of the solvent. Deprotected compound was then dissolved in dry acetonitrile (2 mL) and Boc-GCP-OH (11.0 mg, 0.03 mmol), HBTU (8.5 mg, 0.02 mmol, 98%), HOBT (3.0 mg, 0.02 mmol, 97%) and Et3N (11.2 μL, 0.08 mmol) were added. Reaction was stirred at room temperature overnight. Product 4-Boc was isolated by preparative thin layer chromatography in dichloromethane:methanol 9:1 (slightly yellow solid, 7.3 mg, 58%). To the solution of Boc protected amino acid 4-Boc (20.5 mg, 0.03 mmol) in dichloromethane (4 mL) was added TFA-H2O mixture (9:1, v/v, 2 mL) and the reaction was stirred at room temperature overnight. Solvent was then evaporated and residue purified by preparative thin layer chromatography in dichloromethane:methanol 9:1. Trifluoroacetic salt of deprotected amino acid was obtained as colourless oil, dissolved in methanol (2 mL) and hydrochloric acid (5%, 2 mL) was added. Reaction was stirred at room temperature for 1 hour. Solvent was then evaporated to afford the hydrochloride salt 4 of the deprotected compound as pale yellow solid (18.2 mg, 93%).M.p. > 300 °C (decomposition); IR (KBr) νmax cm−1: 3329 (s), 2978 (m), 2922 (m), 1699 (s), 1655 (s), 1647 (s), 1556 (s), 1475 (m), 1441 (m), 1406 (w), 1364 (w), 1288 (s), 1254 (m), 1202 (m), 1161 (w), 1072 (w), 854 (w), 814 (w), 764 (m), 721 (w), 602 (m); 1H NMR (DMSO-d6) δ/ppm: 12.28 (s, 1H, NH–Pyrr), 12.02 (s, 1H, NH–Guan), 8.95–8.85 (m, 2H, CH–Phen), 8.76–8.42 (m, 7H, NH–Guan, NH2–Guan, NH–Ala, NH–Gly, CH–Phen), 8.37 (bs, 1H, CH–Phen), 8.14 (bs, 1H, CH–Phen), 8.00–7.85 (m, 2H, CH–Phen), 7.48 (s, 1H, CH–Pyrr), 6.75 (s, 1H, CH–Pyrr), 4.81–4.67 (m, 1H, CH–Ala), 3.90–3.77 (m, 2H, CH2–Gly), 3.69 (s, 3H, OCH3), 3.45–3.17 (m, 5H, CH2–Ala, CH3); 13C NMR (DMSO-d6) δ/ppm: 172.45 (Cq), 171.52 (Cq), 168.87 (Cq), 168.71 (Cq), 159.47 (Cq), 159.12 (Cq), 155.44 (Cq), 132.18 (Cq), 131.85 (Cq), 130.29 (CH–Phen), 129.25 (CH–Phen), 128.84 (CH–Phen), 125.42 (Cq), 124.04 (Cq), 123.66 (Cq), 123.42 (CH–Phen), 123.23 (CH–Phen), 115.78 (CH–Pyrr), 112.77 (CH–Pyrr), 52.84 (CH–Ala), 52.08 (OCH3), 41.74 (CH2–Gly), 36.29 (CH2–Ala), 19.45 (q, CH3); HRMS: m/z: calcd for C27H27N7O5+: 530.2146; found 530.2169 [M + H]+.
Molecular simulations
3D structures of ligands C27H25N6O5+ and C25H25N6O4+ denoted as 2 and 3, respectively were constructed using the program GaussView 5.0.20 The ligand docking was performed in two ways: using the program Autodock (AutoDock 4.2.6)21 and manually using and crystal structure of the h.DPP III – tynorphin complex as a templated utilizing the program PyMOL (https://www.pymol.org/).
The complexes were parametrized by the AMBERTools14 modules antechamber and tleap using GAFF22 and ff12SB23 force fields to parameterize the substrate and the protein, respectively. For the zinc cation, Zn2+, parameters were derived from our previous work24 and modified according to the PDB survey.25
The complexes were solvated in an octahedron box filled with TIP3P26 water molecules ensuring a 12 Å thick water molecules buffer around the protein. Na+ ions were added to neutralize the system, and were placed in the vicinity of the charged amino acid residues at the protein surface.
000 atoms (∼28000 molecules of water), were simulated using periodic boundary conditions.27 The electrostatic interactions were calculated using the particle-mesh Ewald method.28 The pmemd module running at GPU was used to conduct the molecular dynamics (MD) simulation. Before MD simulations, the protein geometry was optimized using the 3 module sander in three cycles, each using different constraints on the protein. In the first cycle 1500 optimization steps were preformed, where the first 450 steps were of the steepest descent method, and the rest was the conjugate gradient. Both, the protein atoms and the metal ion, were constrained using a harmonic potential of 32 kcal (mol−1 Ǻ−2), in order to equilibrate water molecules. In the second cycle 2500 steps of optimization was done. The metal ion and the protein backbone were constrained with 12 kcal (mol−1 Ǻ−2). Finally in the third cycle the same number of optimization steps was used as in first cycle, and the protein backbone and the metal ion were constrained with 1 kcal (mol−1·Ǻ−2) and 12 kcal (mol−1·Ǻ−2), respectively. The minimized system was heated from 0 to 300 K during 30 ps using a canonical ensemble (NVT), followed equilibration stage of 30 ps, during which the water density was adjusted. The equilibrated system was then subjected to at least 60.2 ns of the productive MD simulations. In all except one case the following scheme was applied: 20 ns at 300 K, heating from 300 to 400 K during 0.1 ns, 20 ns at 400 K, cooling from 400 to 300 K during 0.1 ns, 20 ns at 300 K. Constant temperature (either 300 or 400 K) and pressure (1 atm) were ensured using Langevin dynamics with a collision frequency of 1 ps−1, and Berendsen barostat, respectively. The time step during the periods of heating and cooling was 1 fs, and for the rest of the simulation 2 fs (to restrain the motion of hydrogens the SHAKE algorithm was used). The 20 ns long periods of simulations at constant temperature were performed using NPT conditions, while during heating and cooling period a canonical ensemble (NVT) was simulated.Acknowledgements
The work has been supported by Croatian Science Foundation projects: 7235 and 1477, as well as by the FP7-REGPOT-2012-2013-1 project, Grant Agreement Number 316289 – InnoMol. We thank Marija Kozlović, M.S., for the purification of human recombinant DPP III, and also Peter Macheroux team (TU-Graz, Austria) for valuable support and discussions.Notes and references
- M. Baršun, N. Jajčanin, B. Vukelić, J. Špoljarić and M. Abramić, Biol. Chem., 2007, 388, 343–348 CrossRef PubMed.
- Y. X. Liu, J. T. Kern, J. R. Walker, J. A. Johnson, P. G. Schultz and H. Luesch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 5205 CrossRef CAS PubMed.
- B. E. Hast, D. Goldfarb, K. M. Mulvaney, M. A. Hast, P. F. Siesser, F. Yan, D. N. Hayes and M. B. Major, Cancer Res., 2013, 73, 2199 CrossRef CAS PubMed.
- A. Tomić, M. Abramić, J. Špoljarić, D. Agić, D. M. Smith and S. Tomić, J. Mol. Recognit., 2011, 24, 804 CrossRef PubMed.
- J. Špoljarić, A. Tomić, B. Vukelić, B. Salopek-Sondi, D. Agić, S. Tomić and M. Abramić, Croat. Chem. Acta, 2011, 84, 259 CrossRef.
- A. Tomić, M. Gonzalez and S. Tomić, J. Chem. Inf. Model., 2012, 52, 1583 CrossRef PubMed.
- A. Tomić, M. Berynskyy, R. C. Wade and S. Tomić, Mol. BioSyst., 2015, 11, 3068 RSC.
- A. Tomić and S. Tomić, Dalton Trans., 2014, 43, 15503 RSC.
- J. Špoljarić, B. Salopek-Sondi, J. Makarević, B. Vukelić, D. Agić, S. Šimaga, N. Jajčanin-Jozić and M. Abramić, Bioorg. Chem., 2009, 37, 70 CrossRef PubMed.
- A. Cvitešić, I. Sabljić, J. Makarević and M. Abramić, J. Enzyme Inhib. Med. Chem. DOI:10.1080/14756366.2016.1186021.
- C. Schmuck, Synlett, 2011, 1798 CrossRef CAS.
- S. Langolf, U. Machon, M. Ehlers, W. Sicking, T. Schirmeister, C. Buchhold, C. Gelhaus, P. J. Rosenthal and C. Schmuck, ChemMedChem, 2011, 6, 1581 CrossRef CAS PubMed.
- K. Klemm, M. Radić-Stojković, G. Horvat, V. Tomisić, I. Piantanida and C. Schmuck, Chem.–Eur. J., 2012, 18, 1352 CrossRef CAS PubMed.
- M. Radić-Stojković, P. Piotrowski, C. Schmuck and I. Piantanida, Org. Biomol. Chem., 2015, 13, 1629 Search PubMed.
- M. Dukši, D. Baretić, V. Čaplar and I. Piantanida, Eur. J. Med. Chem., 2010, 45, 2671 CrossRef PubMed.
- G. A. Kriss, Specfit programme for nonlinear least-squares regression of multiwavelength spectrophotometric data, in Astronomical Data Analysis Software & Systems III, A.S.P. Conf. Series, ed. D. R. Crabtree, R. J. Hanisch and J. Barnes, Astronomical Society of the Pacific, San Francisco, 1994, vol. 61, p. 437 Search PubMed.
- E. Dobrovetsky, A. Dong, A. Seitova, B. Duncan, L. Crombet, M. Sundstrom, et al. Crystal structure of human dipeptidyl peptidase III. Structural Genomics Consortium (SGC), 2009.
- G. A. Bezerra, E. Dobrovetsky, R. Viertlmayr, A. Dong, A. Binter, M. Abramić, P. Macheroux, S. Dhe-Paganon and K. Gruber, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 6525 CrossRef CAS PubMed.
- D. A. Case, V. Babin, J. T. Berryman, R. M. Betz, Q. Cai, D. S. Cerutti, T. E. Cheatham III, T. A. Darden, R. E. Duke, H. Gohlke, A. W. Goetz, S. Gusarov, N. Homeyer, P. Janowski, J. Kaus, I. Kolossváry, A. Kovalenko, T. S. Lee, S. LeGrand, T. Luchko, R. Luo, B. Madej, K. M. Merz, F. Paesani, D. R. Roe, A. Roitberg, C. Sagui, R. Salomon-Ferrer, G. Seabra, C. L. Simmerling, W. Smith, J. Swails, R. C. Walker, J. Wang, R. M. Wolf, X. Wu and P. A. Kollman, AMBER 14, University of California, San Francisco, 2014 Search PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, v. 5.0, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785 CrossRef CAS PubMed.
- J. M. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, J. Comput. Chem., 2004, 25, 1157 CrossRef CAS PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1996, 118, 2309 CrossRef CAS.
- B. Bertoša, B. Kojić-Prodić, R. C. Wade and S. Tomić, Biophys. J., 2008, 94, 27 CrossRef PubMed.
- I. Dokmanić, M. Šikić and S. Tomić, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2008, 64, 257 Search PubMed.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926 CrossRef CAS.
- T. Darden, D. Pearlman and L. G. Pedersen, J. Chem. Phys., 1998, 109, 10921 CrossRef CAS.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089 CrossRef CAS.
- P. Kumar, V. Reithofer, M. Reisinger, S. Wallner, T. Pavkov-Keller, P. Macheroux and K. Gruber, Sci. Rep., 2016, 6, 23787 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16966j |
| This journal is © The Royal Society of Chemistry 2016 |