
Feature extraction from resolution perspective for gas chromatography-mass spectrometry datasets†
Abstract
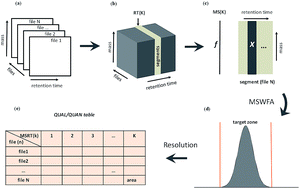
Automatic feature extraction from large-scale datasets is one of the major challenges when analyzing complex samples with gas chromatography-mass spectrometry (GC-MS). The classic processing pipeline basically consists of noise filtering, baseline correction, peak detection, alignment, normalization and identification. The long pipeline makes the extracted features inconsistent with different methods and values of parameters. In this study, MS-Assisted Resolution of Signals (MARS) has been proposed to extract features automatically from resolution perspective for large-scale GC-MS datasets. Firstly, it divides complex data into small segments and searches the target zone by moving sub-window factor analysis (MSWFA). Then, improved iterative target transformation factor analysis (ITTFA) has been developed to extract features of the compound from complex datasets. MARS was systematically tested on a simulated dataset (5 samples), peppermint dataset (2 samples), red wine dataset (24 samples) and human plasma dataset (131 samples). The results show that MARS can extract features accurately, automatically, objectively and swiftly from these complex datasets at 2–3 minutes/chromatogram speed. The extracted features of overlapped peaks are comparable to the features resolved by MCR-ALS or PARAFAC2, and significantly better than XCMS. Furthermore, PLS-DA models of the human plasma dataset indicated that features extracted automatically by MARS are comparable or better than features extracted manually by experts with a GC-MS workstation. It has been implemented and open-sourced at https://github.com/zmzhang/MARS.
 Please wait while we load your content...
Please wait while we load your content...

