
Mono- and triiodophenyl isocyanate as radiopacifying agents for methacrylate-based copolymers; biocompatibility and non-toxicity
Abstract
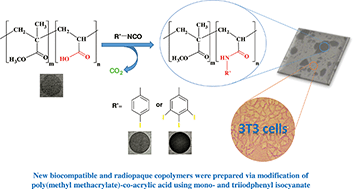
New radiopaque acrylic copolymers were prepared via the copolymerization of methyl methacrylate (MMA) and acrylic acid (AA). The copolymers were made radiopaque through the reaction of carboxylic acid groups with 4-iodophenyl isocyanate and 3,4,5-triiodophenyl isocyanate moieties, as radiopacifying agents. The iodinated copolymers were characterized by Fourier transform infrared (FT-IR) spectroscopy, nuclear magnetic resonance (NMR), energy dispersive X-ray (EDAX), gel permeation chromatography (GPC), elemental analysis, differential scanning calorimetry (DSC), and thermogravimetric analysis (TGA). The composition and iodine content of the copolymers were demonstrated via acid–base and potentiometric titrations, respectively. The radiopacity of the copolymers was investigated by X-radiography. To evaluate the biocompatibility of the iodinated copolymers, a direct MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) assay was carried out on Swiss mouse embryonic stem tissue fibroblast-like cells (NIH3T3 cell line) according to the ISO10993-5 standard. The results indicated that the new iodinated copolymers are thermally stable and have high radiopacity. It was also found that the newly radiopaque copolymers have no cytotoxicity, and could be useful for biomedical applications.
 Please wait while we load your content...
Please wait while we load your content...

