
Two-step three-component process for one-pot synthesis of 8-alkylmercaptocaffeine derivatives†
M. N. Soltani Rad* and
S. Maghsoudi
Medicinal Chemistry Research Laboratory, Department of Chemistry, Shiraz University of Technology, Shiraz 71555-313, Iran. E-mail: soltani@sutech.ac.ir
First published on 19th July 2016
Abstract
A highly efficient, odourless and two-step three-component process for one-pot synthesis of some 8-alkylmercaptocaffeine derivatives has been described. The catalyst-free three-component reaction of alkyl bromides, thiourea, and 8-bromocaffeine gave 8-alkylmercaptocaffeine products in excellent to quantitative yields. In addition, the impact of parameters on sample reaction is discussed.
Introduction
Undoubtedly methylxanthines are one of the most interesting classes of compounds that people around the world are exposed to in their everyday life.1 Methylxanthine alkaloids including caffeine, theophylline, theobromine and their related metabolite i.e. paraxanthine are naturally found in berries, seeds and leaves of many plants like coffee, cocoa and tea (Fig. 1).2 Methylxanthines are unique molecules with immense pharmaceutical activities.3 This is due to the fact that xanthine-based scaffolds are well-recognized by plenty of enzymes or receptors in human and animal cells.4 Methylxanthines and their derivatives have received much attention over the years for their magnificent chemotherapeutic properties as bronchodilator (antiasthmatic, expectorant), antiemetic, cardiotonic, coronary vasodilator, diuretic, analgesic, anti-migraine, anti-rhinitis, analeptic, antispasmodic, psychotonic, CNS stimulant and so on.5 Moreover, methylxanthines are known as antagonizing agents for phosphodiesterase enzymes6 and adenosine receptors.7 Among methylxanthines, most chemical modifications were achieved on theophylline and theobromine.8 Since the presence of nucleophilic nitrogens (i.e.: N7 and N1 in theophylline and theobromine, respectively) they readily prone to react with diverse carbon electrophiles; whereas, the caffeine has neither the intense nucleophilic nor electrophilic sites to react with active chemical species (Fig. 1). In particular, the caffeine can be affected merely from C(8) site through the reaction with certain electrophiles like positive halogens and so on.9 | ||
| Fig. 1 The structures of xanthine and natural methylxanthines. | ||
The structural activity relationship (SAR) studies of natural methylxanthines and their synthetic derivatives have revealed that the site of alkylation almost determines the pharmaceutical and the biological behavior trend of the corresponding methylxanthine.10 As an instance, the C(8) alkylation/substitution of caffeine has often led to the new analogues with potential anti-rhinitis or antagonistic property for adenosine receptors as well as phosphodiesterase enzymes; whereas, the N(1)-alkylation of theobromine shifts the property to vasodilatory activity (Fig. 2).11
 | ||
| Fig. 2 The modifications (alkylation or substitution) to scaffolds of methylxanthines lead to diverse pharmaceutical properties. | ||
Since the discovery of 6-mercaptopurine (6-MP) as an immunosuppressive medication for treating acute lymphocytic leukemia12 and also 8-methylmercaptoxanthine as a main metabolite of oncogene (i.e.: 3-hydroxyxanthine),13 efforts have been made to incorporate the mercapto moiety into scaffolds of diverse methylxanthines to access new bioactive derivatives. In this connection, 8-thiosubstituted methylxanthines especially 8-mercaptocaffeine derivatives have found particular attention since their noticeable pharmaceutical profiles. Up to now, few numbers of these derivatives have been developed. Petzer and coworkers reported the synthesis of 8-[(alkyl) sulfanyl] caffeine derivatives 1 as potent MAO-B inhibitors through C-8 substitution of caffeine with thioether moieties.14,15 Krutovskikh et al. reported the synthesis of 8-β-dialkylaminoethylmercaptocaffeine analogous 2 with radio-protective property.16 Caffeine-8-thioglycolic acid derivatives 3 were synthesized by Zlatkov and coworkers with brain antihypoxic activity.17 In addition, several 8-mercapto-caffeine derivatives and/or related scaffolds 4–8 with certain biological activities have been established.18–22 The structures of compounds 1–8 and their corresponding therapeutic activities are shown in Fig. 3.
 | ||
| Fig. 3 The structures and therapeutic activities of 8-alkylmercaptocaffeine and their related derivatives. | ||
To access 8-alkylmercaptocaffeine derivatives, diverse approaches have been reported so for comprising (i) reaction of 8-mercaptocaffeine [CAS no: 56223-58-6] with alkyl halides in the presence of an efficient base,16,23 (ii) direct thiolation reaction of mercaptans with 8-chlorocaffeine (8-CC) or 8-bromocaffeine (8-BC) in basic media14,15,24 and more recently, (iii) copper-catalyzed direct thiolation of isocaffeine via disulfides utilized AgOAc as an additive under O2 atmosphere.25 Despite the usefulness of the above approaches for synthesis of some 8-alkylmercaptocaffeine derivatives, the drawbacks still have remained. As instances, the reaction of 8-mercaptocaffeine with alkyl halides16,23 requires strong bases such as EtONa which often leads to the low yields of products. The low yield of products is attributed to narrow nucleophilic power of 8-mercaptocaffeine. The weak nucleophilic potency of 8-mercaptocaffeine is owing to the fact that charge density on –SH moiety is distributed between sulfur and adjacent N7 and N9 atoms through the possible tautomerization effect.23 Therefore, the application scope of S-alkylation reaction for 8-mercaptocaffeine via alkyl halides is very limited. The direct thiolation reaction of mercaptans with 8-chlorocaffeine (8-CC) or 8-bromocaffeine (8-BC) in basic media also associates with two main drawbacks involving the extreme of unpleasant and disgusting odours of used mercaptans, and sensitivity of mercaptans toward oxidizing agents like atmospheric oxygen which readily converts the thiols into disulfides at basic pH (pH > 7). Moreover, an approach developed by Zhu and coworkers25 for straightforward thiolation of isocaffeine requires less available and presynthesized disulfides, the use of an expensive additive and performing the reaction under an O2 atmosphere which is not simple and easy to handle.
Thiourea is a safe, cheap and widely available organosulfur compound which has been extensively used for generation of thiols from alkyl halides.26,27 The major advantages associated with the use of thiourea is the in situ generation of thiols. The in situ generated thiols subsequently can be used to react with set of diverse carbon electrophiles in one-pot reaction condition to access a plenty of thio-compounds. This advantage prevents the requirement of extra procedure for separation and purification of thiols and thus diminishes the intense of the odd smell of thiols and largely reduces the oxidative homocoupling of thiols to disulfides.28–30
Regarding to remarkable therapeutic activity of 8-alkylmercaptocaffeine derivatives and also in continuation of our long-standing interest on synthesis of new methylxanthine derivatives;8,31 hereby, we would like to report highly efficient, catalyst-free, odourless and three-component process for one-pot synthesis of some 8-alkylmercaptocaffeine (Scheme 1).
 | ||
| Scheme 1 Synthesis of 8-alkylmercaptocaffeine through one-pot, three-component reaction using thiourea, alkyl bromide and 8-BC. | ||
In this synthesis, the reaction of thiourea with alkyl bromides in refluxing EtOH followed by addition of aqueous NaOH at room temperature provided the corresponding 8-alkylmercaptocaffeine derivatives 12a–q in excellent yields.
Results and discussion
To start synthesizing the title compounds, we first considered the synthesis of 12k as a sample compound as this potent inhibitor of MAO-B was previously synthesized by Petzer et al.14,15 We initially attempted to synthesize 12k through the reaction of 8-mercaptocaffeine with phenethyl bromide in refluxing absolute ethanol. However, no reaction was carried out after refluxing for 48 h. Additionally, utilizing organic and inorganic bases to improve the reaction yield were only acquired the moderate yield of 12k. As an instance, the use of fresh EtONa in absolute ethanol only afforded 12k in 45% yield after prolonging the reaction time up to 72 h. As mentioned earlier, the low yield of product obtained by direct reaction of 8-mercaptocaffeine with alkyl halides is caused by weak nucleophilic nature of 8-mercaptocaffeine.8-Bromo-(8-BC) or 8-chlorocaffeine (8-CC) are known as interesting compounds for introduction of 8-caffeinyl moiety to diverse nucleophiles.14,15,17,24,32 As matter of fact, the linkage of C(8) with three electron withdrawing elements in 8-BC or 8-CC extensively enhances the positive charge density on C(8) which dispose to be attacked by varied nucleophiles through SNAr-type reaction. Thus, to obtain the higher yield of 12k, an alternative approach was applied in which 2-phenylethanethiol (CAS no: 4410-99-5) was directly reacts with 8-BC in the presence of an efficient base (NaOH) in EtOH at reflux condition due to procedure reported by Petzer et al.14,15 and Long.24 Although a good yield of 12k (>78%) were obtained using current approach, this procedure suffer from the extreme of unpleasant odours of the used mercaptan.
Regarding to numerous benefits of one-pot multicomponent reactions (MCR),33 we applied this strategy to overcome the addressed issues. To this end, the one-pot two-step three component reaction of thiourea as a sulfur source agent with alkyl halides followed by addition of 8-BC seems to be an attractive and effectual strategy to access 8-alkylmercaptocaffeine derivatives.
To find out the optimized reaction conditions, we screened the influence of parameters like solvent and base. In this connection, the optimization was begun with studying the influence of various aprotic, protic and other solvents on the sample reaction in the presence of aqueous NaOH as a base (Table 1). As shown in Table 1, through the examined solvents, ethanol (Table 1, entry 10) afforded the best result and hence it was solvent of choice for all next reactions. In general, the use of protic solvents (Table 1, entries 10–13) gained the better results compared to aprotic solvents while in contrary, water (Table 1, entry 9) was insufficient to process the reaction. In addition to ethanol, methanol also afforded an excellent yield of 12k; however, since its toxicity, the use of ethanol was preferred. Utilizing polar aprotic solvents (Table 1, entries 1–7) acquired low to moderate yields of 12k in different times. Whereas, toluene and a room temperature ionic liquid (i.e.: bmim[Br]) were failed to improve the reaction (Table 1, entries 8 and 14).
|
||||
|---|---|---|---|---|
| Entry | Solvent | Baseb | Time (h) | Yieldc(%) |
| a Reaction conditions: phenethyl bromide (10 mmol), thiourea (12 mmol), 8-BC (10 mmol), solvent (25 mL), base (20 mmol) and H2O (2 mL).b Except that of H2O as a solvent (entry 9), NaOH was primarily dissolved in H2O (2 mL) and then was added to reaction mixture.c Isolated yield.d The reaction was conducted at 100 °C.e No reaction. | ||||
| 1 | DMFd | NaOH | 34 | 42 |
| 2 | THF | NaOH | 40 | 38 |
| 3 | DMSOd | NaOH | 37 | 40 |
| 4 | MeCN | NaOH | 42 | 32 |
| 5 | NMPd | NaOH | 44 | 38 |
| 6 | HMPAd | NaOH | 48 | 32 |
| 7 | Me2CO | NaOH | 48 | 29 |
| 8 | PhMe | NaOH | 72 | NRe |
| 9 | H2O | NaOH | 72 | 25 |
| 10 | EtOH | NaOH | 14 | 90 |
| 11 | i-PrOH | NaOH | 22 | 75 |
| 12 | MeOH | NaOH | 17 | 86 |
| 13 | PEG 200d | NaOH | 36 | 65 |
| 14 | bmim[Br]d | NaOH | 72 | NR |
| 15 | EtOH | — | 72 | NR |
| 16 | EtOH | TEA | 48 | 36 |
| 17 | EtOH | K2CO3 | 72 | NR |
| 18 | EtOH | DBU | 28 | 38 |
| 19 | EtOH | MgO | 72 | NR |
| 20 | EtOH | EtONa | 18 | 40 |
| 21 | EtOH | t-BuOK | 24 | 35 |
| 22 | EtOH | DBN | 30 | 39 |
| 23 | EtOH | DMAP | 26 | 31 |
| 24 | EtOH | DABCO | 36 | 29 |

Afterward of determining a proper solvent, we then examined the influence of diverse organic and inorganic bases. The use of an efficient base has undeniable role in progress of reaction. In absence of a base, the reaction was not conducted at all even if the reaction time has been extended to three days and more (Table 1, entry 15). As can be seen in Table 1, none of tested bases were proper for progress of reaction. The only base that found suitable for this synthesis was aqueous NaOH which employed for all reactions.
With the optimal reaction conditions in hand, we screened the versatility and the scope of this current approach. In this regard, a variety of alkyl bromides were proved to be effective to generate products 12 in excellent to almost quantitative yields (Table 2).











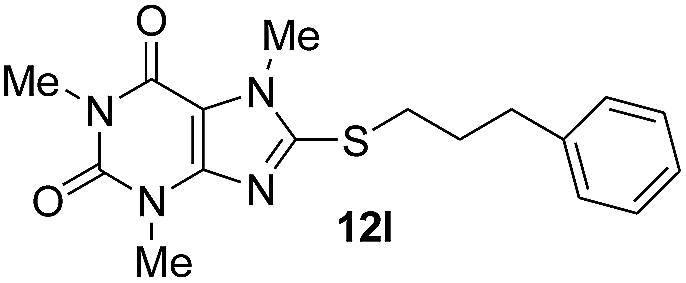





The alkyl bromides with different normal carbon chains readily underwent the reactions with thiourea and 8-BC in excellent yields (Table 2, entries 1–7). Interestingly, as expected, among tested alkyl bromides, the maximum yields were obtained for alkyl bromides with shorter normal carbon chains; whereas, the yields were gradually diminished by increasing the number of carbons in a chain. Also the utilizing alkyl bromide with a branched aliphatic chain acquired a good yield of product (Table 2, entry 8). Moreover, alkyl bromides with ω-substituted phenyl moieties (Table 2, entries 11 and 12) were afforded the corresponding products 12k and 12l in excellent yields. This current approach also works well with benzyl bromide derivatives in which 9i, 9j, 9m and benzhydryl bromide were efficiently converted to the corresponding products also in quantitative yields. In addition, alkyl bromides with N-heterocyclic compounds at ω-position involving benzimidazolyl (9o), 2-methyl-4-nitroimidazolyl (9p) and phthalimidyl (9q) were almost quantitatively converted to corresponding 8-alkylmercapto-caffeine (12o–12q). It is worth mentioning that the other alkyl halides involving alkyl chlorides and iodides along with alkyl sulfonates could be applied; however, to our experience the best results were obtained when alkyl bromides were employed.
On the basis of the experimental results, a plausible mechanism for the formation of products is proposed (Scheme 2). As shown in Scheme 2, the thiourea first performs the nucleophilic attack to alkyl bromide to generate alkylthiouronium hydrobromide salt (I). The generation of this salt was obviously observed during the reaction course. Afterward, salt (I) is hydrolyzed to urea and corresponding thiol (II) by addition of hydroxide ion. The in situ generated thiol (II) is then activated by extra hydroxide ion and attacks to 8-BC through SNAr-type reaction to afford 8-alkylmercaptocaffeine.
 | ||
| Scheme 2 A plausible mechanism for synthesis of 8-alkylmercaptocaffeine using thiourea, alkyl bromide and 8-BC. | ||
Conclusions
In conclusion, we have developed an efficient, odourless and practical route for one-pot two-step three-component synthesis of some 8-alkylmercaptocaffeine derivatives. The three-component reaction of alkyl bromides, thiourea and 8-BC in the presence of aqueous NaOH in ethanol acquired 8-alkylmercaptocaffeine derivatives in excellent to quantitative yields. The influence of parameters effective on reaction progress involving solvent and base effects has been studied. This catalyst-free approach proceeds smoothly under mild conditions from readily available starting materials and provides diverse 8-alkylmercaptocaffeine as potential chemotherapeutic agents.Experimental
All chemical reagents except that of 8-bromocaffeine (8-BC)34 were purchased from Fluka, Sigma-Aldrich or Merck companies. Solvents were purified by standard procedures, and stored over 3 Å molecular sieves.35 Reactions were followed by TLC using SILG/UV 254 silica-gel plates. Column chromatography was performed on silica gel 60 (0.063–0.200 mm, 70–230 mesh; ASTM). IR spectra were obtained using a Shimadzu FT-IR-8300 spectrophotometer. 1H and 13C-NMR spectrum was recorded on Brüker Avance-DPX-250 spectrometer operating at 250/62.5 MHz, respectively. Chemical shifts are given in δ relative to tetramethylsilane (TMS) as an internal standard, coupling constants J are given in Hz. Abbreviations used for 1H NMR signals are: s = singlet, d = doublet, t = triplet, q = quartet, m = multiplet, br = broad and etc. GC/MS was performed on a Shimadzu GC/MS-QP 1000-EX apparatus (m/z; rel.%). Elemental analyses were performed on a Perkin-Elmer 240-B micro-analyser.General procedure for synthesis of 8-alkylmercaptocaffeine (12a–12q)
In a triple-necked round bottom flask (100 mL) equipped with a condenser and additional funnel, it was added a mixture, consisting of alkyl bromide (10 mmol) and thiourea (12 mmol, 0.91 g) in 96% EtOH (25 mL). The mixture was heated at reflux and heating was continued until TLC indicated the completion of reaction. Afterward, the solution of NaOH (0.02 mol, 0.8 g) in distillated water (2 mL) was added to reaction mixture through the connected addition funnel at reflux for an extra hour. The reaction media was chilled to room temperature and then 8-BC (10 mmol, 2.73 g) was added and the mixture was stirred at room temperature. In most cases, the suspension of 8-BC crystals immediately vanished which is a good evidence for rapid progression of reaction (note: 8-BC is not dissolved in EtOH at room temperature). After completion of the reaction (TLC check), the solvent was evaporated in vacuo and the remaining residue was diluted in CHCl3 (100 mL) and subsequently washed with water (2 × 100 mL). The organic layer was dried (Na2SO4) and evaporated. The crude product was purified by recrystallization and/or column chromatography on silica gel eluting with proper solvents described below.36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6).
:6).IR (KBr): 2952, 2832, 1701, 1658, 1583, 1447, 1341 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.84 (s, 3H, N(7)–CH3), 3.55 (s, 3H, N(1)–CH3), 3.38 (s, 3H, N(3)–CH3), 3.25 (t, J = 7.2 Hz, 2H, SCH2), 1.86–1.71 (m, 2H, SCH2CH2), 1.05 (t, J = 7.3 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 155.1, 152.2, 151.3, 150.4, 105.1, 38.7, 33.5, 30.0, 24.8, 21.3, 13.8.
MS (EI): m/z (%) = 268 (15.7) [M+].
Anal. calcd for C11H16N4O2S: C, 49.24; H, 6.01; N, 20.88. Found: C, 49.32; H, 6.15; N, 20.96.
:6).IR (KBr): 2964, 2873, 1701, 1671, 1560, 1426, 1372 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.69 (s, 3H, N(7)–CH3), 3.46 (s, 3H, N(1)–CH3), 3.32 (s, 3H, N(3)–CH3), 3.17 (t, J = 7.1 Hz, 2H, SCH2), 1.52–1.20 (complex, 4H, 2 CH2), 0.88 (t, J = 7.1 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 155.8, 152.8, 151.8, 150.6, 107.3, 37.2, 33.2, 32.6, 29.6, 22.0, 21.7, 13.8.
MS (EI): m/z (%) = 282 (13.4) [M+].
Anal. calcd for C12H18N4O2S: C, 51.04; H, 6.43; N, 19.84. Found: C, 51.17; H, 6.51; N, 19.76.
:15) afforded a white solid; yield: 2.66 g (90%); mp 75–77 °C; Rf = 0.36 (EtOAc–n-hexane, 1:6).IR (KBr): 2946, 2847, 1702, 1663, 1538, 1444, 1349 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.82 (s, 3H, N(7)–CH3), 3.53 (s, 3H, N(1)–CH3), 3.36 (s, 3H, N(3)–CH3), 3.26 (t, J = 7.3 Hz, 2H, SCH2), 1.82–1.70 (m, 2H, SCH2CH2), 1.49–1.30 (m, 4H, 2 CH2), 0.92 (t, J = 7.0 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 154.9, 151.9, 150.6, 149.1, 108.8, 36.9, 32.3, 31.0, 30.1, 29.5, 22.7, 21.8, 14.4.
MS (EI): m/z (%) = 296 (18.9) [M+].
Anal. calcd for C13H20N4O2S: C, 52.68; H, 6.80; N, 18.90. Found: C, 52.83; H, 6.87; N, 19.02.
:15) afforded a white solid; yield: 2.79 g (90%); mp 67–69 °C; Rf = 0.35 (EtOAc–n-hexane, 1:6).IR (KBr): 2942, 2910, 1710, 1652, 1551, 1478, 1362 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.73 (s, 3H, N(7)–CH3), 3.44 (s, 3H, N(1)–CH3), 3.26 (s, 3H, N(3)–CH3), 3.17 (t, J = 7.3 Hz, 2H, SCH2), 1.72–1.60 (m, 2H, SCH2CH2), 1.38–1.22 (complex, 6H, 3 CH2), 0.78 (t, J = 7.3 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 155.6, 152.0, 151.1, 149.7, 106.5, 37.1, 33.3, 31.8, 30.6, 29.1, 28.5, 22.9, 21.7, 15.8.
MS (EI): m/z (%) = 310 (17.5) [M+].
Anal. calcd for C14H22N4O2S: C, 54.17; H, 7.14; N, 18.05. Found: C, 54.09; H, 7.21; N, 18.13.
:6).IR (KBr): 2943, 2863, 1703, 1682, 1579, 1431, 1388 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.84 (s, 3H, N(7)–CH3), 3.55 (s, 3H, N(1)–CH3), 3.38 (s, 3H, N(3)–CH3), 3.26 (t, J = 7.3 Hz, 2H, SCH2), 1.80–1.72 (m, 2H, SCH2CH2), 1.46–1.29 (complex, 8H, 4 CH2), 0.88 (t, J = 6.8 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 156.2, 152.9, 151.4, 150.2, 108.9, 37.0, 32.4, 31.8, 30.6, 29.7, 28.8, 27.9, 22.5, 21.8, 14.3.
MS (EI): m/z (%) = 324 (19.3) [M+].
Anal. calcd for C15H24N4O2S: C, 55.53; H, 7.46; N, 17.27. Found: C, 55.61; H, 7.40; N, 17.35.
:15) afforded a white solid; yield: 2.87 g (85%); mp 66–68 °C; Rf = 0.34 (EtOAc–n-hexane, 1:6).IR (KBr): 2945, 2852, 1702, 1672, 1586, 1463, 1357 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.76 (s, 3H, N(7)–CH3), 3.47 (s, 3H, N(1)–CH3), 3.30 (s, 3H, N(3)–CH3), 3.18 (t, J = 7.3 Hz, 2H, SCH2), 1.72–1.61 (m, 2H, SCH2CH2), 1.36–1.20 (complex, 10H, 5 CH2), 0.77 (t, J = 6.8 Hz, 3H, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 154.8, 152.1, 151.8, 149.4, 105.1, 37.0, 32.6, 31.6, 30.8, 30.5, 29.9, 29.6, 28.7, 22.5, 20.4, 15.3.
MS (EI): m/z (%) = 338 (20.6) [M+].
Anal. calcd for C16H26N4O2S: C, 56.78; H, 7.74; N, 16.55. Found: C, 56.88; H, 7.81; N, 16.47.
:6).IR (KBr): 2986, 2880, 1706, 1674, 1536, 1472, 1313 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.90 (s, 3H, N(7)–CH3), 3.62 (s, 3H, N(1)–CH3), 3.46 (s, 3H, N(3)–CH3), 3.29 (t, J = 7.3 Hz, 2H, SCH2), 1.74–1.21 (complex, 19H, 8 CH2, CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 155.4, 151.1, 150.5, 149.1, 105.0, 36.9, 33.1, 32.0, 30.6, 30.0, 29.1, 28.8, 28.6, 28.0, 27.4, 21.7, 20.2, 12.5.
MS (EI): m/z (%) = 366 (23.8) [M+].
Anal. calcd for C18H30N4O2S: C, 58.98; H, 8.25; N, 15.29. Found: C, 59.05; H, 8.36; N, 15.38.
:6).IR (KBr): 2956, 2864, 1710, 1678, 1557, 1468, 1369 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 3.76 (s, 3H, N(7)–CH3), 3.47 (s, 3H, N(1)–CH3), 3.31 (s, 3H, N(3)–CH3), 3.19 (t, J = 7.4 Hz, 2H, SCH2), 1.71–1.51 (m, 3H, CH2CH), 0.89 (d, J = 6.3 Hz, 6H, 2 CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 155.4, 152.4, 151.5, 150.5, 106.1, 37.3, 34.2, 31.5, 29.3, 26.9, 22.0, 20.5.
MS (EI): m/z (%) = 296 (17.6) [M+].
Anal. calcd for C13H20N4O2S: C, 52.68; H, 6.80; N, 18.90. Found: C, 52.82; H, 6.87; N, 18.95.
:15) afforded a white solid; yield: 3.17 g (96%); mp 75–77 °C; Rf = 0.28 (EtOAc–n-hexane, 1:6).IR (KBr): 3028, 2987, 2931, 1698, 1674, 1596, 1469, 1368 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 7.28 (d, J = 8.0 Hz, 2H, aryl), 7.16 (d, J = 7.9 Hz, 2H, aryl), 4.45 (s, 2H, SCH2), 3.76 (s, 3H, N(7)–CH3), 3.63 (s, 3H, N(1)–CH3), 3.42 (s, 3H, N(3)–CH3), 2.36 (s, 3H, PhCH3).
13C NMR (CDCl3, 62.5 MHz): δ = 154.4, 151.8, 150.9, 150.3, 137.0, 136.6, 129.8, 127.2, 107.3, 39.1, 32.6, 30.0, 25.2, 22.2.
MS (EI): m/z (%) = 330 (14.6) [M+].
Anal. calcd for C16H18N4O2S: C, 58.16; H, 5.49; N, 16.96. Found: C, 58.29; H, 5.40; N, 16.85.
:15) afforded a white solid; yield: 2.97 g (94%); mp 74–76 °C; Rf = 0.26 (EtOAc–n-hexane, 1:6).IR (KBr): 3030, 2946, 2895, 1711, 1671, 1544, 1436, 1310 cm−1.
1H NMR (DMSO-d6, 250 MHz): δ = 7.45–7.31 (m, 5H, aryl), 4.49 (s, 2H, SCH2), 3.63 (s, 3H, N(7)–CH3), 3.41 (s, 3H, N(1)–CH3), 3.17 (s, 3H, N(3)–CH3).
13C NMR (DMSO-d6, 62.5 MHz): δ = 155.1, 152.4, 151.8, 149.4, 138.6, 128.7, 128.4, 127.3, 104.8, 37.4, 31.4, 30.5, 22.1.
MS (EI): m/z (%) = 316 (20.1) [M+].
Anal. calcd for C15H16N4O2S: C, 56.94; H, 5.10; N, 17.71. Found: C, 57.03; H, 5.21; N, 17.79.
:8) afforded a white solid; yield: 2.97 g (90%); mp 71–73 °C; Rf = 0.23 (EtOAc–n-hexane, 1:6).IR (KBr): 3032, 2941, 1719, 1668, 1542, 1456, 1372 cm−1.
1H NMR (DMSO-d6, 250 MHz): δ = 7.29–7.14 (m, 5H, aryl), 3.62 (s, 3H, N(7)–CH3), 3.44 (t, J = 7.2 Hz, 2H, SCH2), 3.34 (s, 3H, N(1)–CH3), 3.13 (s, 3H, N(3)–CH3), 2.97 (t, J = 7.1 Hz, 2H, PhCH2).
13C NMR (DMSO-d6, 62.5 MHz): δ = 157.6, 152.7, 151.8, 150.3, 139.6, 128.7, 127.5, 125.9, 105.3, 38.3, 36.5, 33.1, 30.7, 22.5.
MS (EI): m/z (%) = 330 (22.6) [M+].
Anal. calcd for C16H18N4O2S: C, 58.16; H, 5.49; N, 16.96. Found: C, 58.27; H, 5.55; N, 16.90.
:10) afforded a white solid; yield: 2.86 g (83%); mp 86–88 °C; Rf = 0.23 (EtOAc–n-hexane, 1:6).IR (KBr): 3050, 2934, 2843, 1701, 1661, 1544, 1431, 1376 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 7.32–7.17 (m, 5H, aryl), 3.82 (s, 3H, N(7)–CH3), 3.51 (s, 3H, N(1)–CH3), 3.37 (s, 3H, N(3)–CH3), 3.26 (t, J = 7.0 Hz, 2H, SCH2), 2.77 (t, J = 7.3 Hz, 2H, PhCH2), 2.16–2.03 (m, 2H, SCH2CH2).
13C NMR (CDCl3, 62.5 MHz): δ = 156.3, 153.3, 152.1, 151.6, 139.7, 129.3, 128.0, 126.3, 107.9, 36.1, 35.8, 35.2, 33.7, 30.1, 21.8.
MS (EI): m/z (%) = 344 (21.8) [M+].
Anal. calcd for C17H20N4O2S: C, 59.28; H, 5.85; N, 16.27. Found: C, 59.41; H, 5.94; N, 16.14.
:10) afforded a white solid; yield: 3.40 g (93%); mp 126–128 °C; Rf = 0.23 (EtOAc–n-hexane, 1:6).IR (KBr): 3037, 2934, 2843, 1701, 1661, 1542, 1431, 1376 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 8.03 (d, J = 7.9 Hz, 1H, aryl), 7.83–7.68 (m, 2H, aryl), 7.52–7.27 (m, 4H, aryl), 4.86 (s, 2H, SCH2), 3.57 (s, 3H, N(7)–CH3), 3.54 (s, 3H, N(1)–CH3), 3.31 (s, 3H, N(3)–CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 156.9, 152.4, 151.2, 150.9, 134.2, 133.0, 132.1, 129.3, 128.8, 128.2, 127.9, 127.3, 126.1, 125.2, 106.4, 39.5, 33.5, 30.8, 23.1.
MS (EI): m/z (%) = 366 (18.3) [M+].
Anal. calcd for C19H18N4O2S: C, 62.28; H, 4.95; N, 15.29. Found: C, 62.19; H, 5.01; N, 15.22.
:15) afforded a white solid; yield: 3.1 g (80%); mp 74–76 °C; Rf = 0.22 (EtOAc–n-hexane, 1:6).IR (KBr): 3025, 2964, 1701, 1671, 1660, 1426, 1372 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 7.43–7.40 (m, 4H, aryl), 7.34–7.24 (m, 6H, aryl), 6.17 (s, 1H, SCH), 3.71 (s, 3H, N(7)–CH3), 3.53 (s, 3H, N(1)–CH3), 3.34 (s, 3H, N(3)–CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 156.7, 152.4, 151.6, 150.6, 142.8, 129.5, 128.6, 127.2, 105.5, 51.3, 33.9, 30.2, 22.6.
MS (EI): m/z (%) = 392 (25.4) [M+].
Anal. calcd for C21H20N4O2S: C, 64.27; H, 5.14; N, 14.28. Found: C, 64.39; H, 5.23; N, 14.35.
:1) afforded a pale-yellow foam; yield: 3.53 g (92%); Rf = 0.38 (EtOAc–n-hexane, 4:1).IR (KBr): 3050, 2991, 2876, 1715, 1678, 1537, 1464 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 8.25 (s, 1H, C(2)-H, benzimidazole), 7.77–7.74 (m, 2H, aryl), 7.66–7.62 (m, 2H, aryl), 3.71 (t, J = 6.8 Hz, 2H, NCH2), 3.62 (s, 3H, N(7)–CH3), 3.41 (s, 3H, N(1)–CH3), 3.27 (s, 3H, N(3)–CH3), 3.06 (t, J = 6.8 Hz, 2H, SCH2), 2.34–2.25 (m, 2H, NCH2CH2).
13C NMR (CDCl3, 62.5 MHz): δ = 155.5, 152.5, 152.1, 151.0, 144.9, 139.1, 134.6, 123.6, 123.0, 115.7, 115.4, 107.0, 54.8, 35.1, 32.9, 31.7, 29.6, 21.7.
MS (EI): m/z (%) = 384 (21.6) [M+].
Anal. calcd for C18H20N6O2S: C, 56.23; H, 5.24; N, 21.86. Found: C, 56.35; H, 5.32; N, 21.78.
:1) afforded a yellow oil; yield: 3.79 g (90%); Rf = 0.36 (EtOAc–n-hexane, 4:1).IR (film): 3047, 2965, 2861, 1718, 1680, 1552, 1534, 1473, 1348 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 7.65 (s, 1H, C(5)-H, imidazole), 3.86 (t, J = 7.0 Hz, 2H, NCH2), 3.64 (s, 3H, N(7)–CH3), 3.44 (s, 3H, N(1)–CH3), 3.29 (s, 3H, N(3)–CH3), 3.13 (t, J = 7.0 Hz, 2H, SCH2), 1.79–1.73 (m, 2H, CH2), 1.49–1.43 (m, 2H, CH2), 1.34–1.28 (m, 2H, CH2).
13C NMR (CDCl3, 62.5 MHz): δ = 155.9, 152.9, 152.6, 151.7, 151.1, 139.0, 132.5, 106.5, 46.0, 36.8, 33.3, 31.2, 30.9, 29.4, 27.7, 22.4, 12.6.
MS (EI): m/z (%) = 421 (20.3) [M+].
Anal. calcd for C17H23N7O4S: C, 48.44; H, 5.50; N, 23.26. Found: C, 48.35; H, 5.41; N, 23.34.
:1) afforded a creamy foam; yield: 3.67 g (92%); Rf = 0.49 (EtOAc–n-hexane, 4:1).IR (KBr): 3068, 2971, 2879, 1716, 1697, 1662, 1541, 1460 cm−1.
1H NMR (CDCl3, 250 MHz): δ = 7.18–7.13 (m, 2H, aryl), 6.99–6.92 (m, 2H, aryl), 3.77 (s, 3H, N(7)–CH3), 3.43–3.30 (complex, 10H, NCH2, SCH2, N(1)–CH3, N(3)–CH3).
13C NMR (CDCl3, 62.5 MHz): δ = 168.4, 155.3, 152.1, 151.9, 151.0, 133.5, 132.4, 127.7, 107.1, 35.6, 33.0, 30.4, 29.8, 22.3.
MS (EI): m/z (%) = 399 (21.2) [M+].
Anal. calcd for C18H17N5O4S: C, 54.13; H, 4.29; N, 17.53. Found: C, 54.21; H, 4.40; N, 17.65.
Acknowledgements
The authors wish to thank Shiraz University of Technology research council for partial support of this work.Notes and references
- R. Franco, A. Oñatibia-Astibia and E. Martínez-Pinilla, Nutrients, 2013, 5, 4159 CrossRef CAS PubMed.
- B. B. Fredholm, Methylxanthines, Handbook of Experimental Pharmacology, Springer-Verlag Berlin Heidelberg, 2011, vol. 200 Search PubMed.
- Ullmann's Encyclopedia of Industrial Chemistry, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, 7th edn, 2005 Search PubMed.
- B. G. Katzung, S. B. Masters and A. T. Trevor, Basic and Clinical Pharmacology, Lange Basic Science, Mc Graw Hill, New York, 12th edn, 2012 Search PubMed.
- A. Kleeman, J. Engel, B. Kutscher and D. Reichert, in Pharmaceutical Substances, Thieme, Stuttgart, 3rd edn, 1999 Search PubMed.
- V. Boswell-Smith, D. Spina and C. P. Page, Br. J. Pharmacol., 2006, 147, 252 CrossRef PubMed.
- M. Williams and M. F. Jarvis, Pharmacol., Biochem. Behav., 1988, 29, 433 CrossRef CAS.
- M. N. Soltani Rad, S. Behrouz and H. Najafi, Synthesis, 2014, 46, 1380 CrossRef.
- L. Zhang and Y. J. Zhang, Tetrahedron Lett., 2006, 47, 775 CrossRef CAS.
- C. O. Wilson, O. Gisvold and J. H. Block, in Wilson and Gisvold's Textbook of Organic Medicinal and Pharmaceutical Chemistry, ed. J. H. Block and J. M. Beale Jr, Lippincott Williams & Wilkins, Philadelphia, 11th edn, 2004 Search PubMed.
- S. Rivara, G. Piersanti, F. Bartoccini, G. Diamantini, D. Pala, T. Riccioni, M. Antonietta Stasi, W. Cabri, F. Borsini, M. Mor, G. Tarzia and P. Minetti, J. Med. Chem., 2013, 56, 1247 CrossRef CAS PubMed.
- L. Lennard, Eur. J. Clin. Pharmacol., 1992, 43, 329 CrossRef CAS PubMed.
- G. B. Brown, M. N. Teller, I. Smullyan, N. J. M. Birdsall, T.-C. Lee, J. C. Parham and G. Stöhrer, Cancer Res., 1973, 33, 1113 CAS.
- S. Mostert, W. Mentz, A. Petzer, J. J. Bergh and J. P. Petzer, Bioorg. Med. Chem., 2012, 20, 7040 CrossRef CAS PubMed.
- H. P. Booysen, C. Moraal, G. T. Blanche, A. Petzer, J. J. Bergh and J. P. Petzer, Bioorg. Med. Chem., 2011, 19, 7507 CrossRef CAS PubMed.
- G. N. Krutovskikh, M. B. Kolesova, A. M. Rusanov, L. P. Vartanyan and M. G. Shagoyan, Pharm. Chem. J., 1975, 9, 234 CrossRef.
- J. Mitkov, N. Danchev, I. Nikolova and A. Zlatkov, Acta Pharm., 2007, 57, 361 CrossRef CAS PubMed.
- L. K. Abdulrahman, M. M. Al-Mousilly, T. S. Al-Halaibeh and K. M. Al-Azzawii, Int. Res. J. Pharm., 2012, 3, 83 CAS.
- C. Fraire, M. Bani, E. Vanotti and V. Olgiati, US Pat., No: 5470858, 1995.
- B. Fischer, R. Yefidoff, D. T. Major, I. Rutman-Halili, V. Shneyvays, T. Zinman, K. A. Jacobson and A. Shainberg, J. Med. Chem., 1999, 42, 2685 CrossRef CAS PubMed.
- A. D. Settimo, A. M. Marini, G. Primofiore, F. D. Settimo and D. J. Bertini, J. Heterocycl. Chem., 1998, 35, 57 CrossRef.
- J. Neyts, E. De Clercq, R. Singha, Y. H. Chang, A. R. Das, S. K. Chakraborty, S. C. Hong, S.-C. Tsay, M.-H. Hsu and J. R. Hwu, J. Med. Chem., 2009, 52, 1486 CrossRef CAS PubMed.
- S. N. Garmash, N. V. Koval, B. A. Priimenko, N. A. Klyuev and E. A. Skulskaya, Chem. Heterocycl. Compd., 1987, 23, 1228 CrossRef.
- L. M. Long, J. Am. Chem. Soc., 1947, 69, 2939 CrossRef CAS PubMed.
- Z. He, F. Luo, Y. Li and G. Zhu, Tetrahedron Lett., 2013, 75, 5907 CrossRef.
- A. J. Speziale, Org. Synth., 1950, 30, 35 CrossRef CAS.
- R. E. Gerber, C. Hasbun, L. G. Dubenko, M. F. King and D. E. Bierer, Org. Synth., 2000, 77, 186 CrossRef CAS.
- G.-p. Lu and C. Cai, RSC Adv., 2014, 4, 59990 RSC.
- N. Azizi, Z. Yadollahy and A. Rahimzadeh-oskooee, Synlett, 2014, 25, 1085 CrossRef.
- H. Firouzabadi, N. Iranpoor and M. Gholinejad, Adv. Synth. Catal., 2010, 352, 119 CrossRef CAS.
- M. N. Soltani Rad, Ph.D. Dissertation, Shiraz University, Shiraz, Iran, October 2004.
- M. N. Soltani Rad, S. Behrouz and A. R. Nekoei, Synlett, 2012, 23, 1191 CrossRef.
- B. B. Toure and D. G. Hall, Chem. Rev., 2009, 109, 4439 CrossRef CAS PubMed.
- 8-Bromocaffeine (CAS no: 10381-82-5) is commercially available material; however, the use of fresh 8-BC in reaction gains the better result. The fresh 8-BC can be prepared due to our more safe established procedure (see ref. 32) or other literature (see ref. 17).
- A. I. Vogel, in Practical Organic Chemistry, Longmans, Green, London, England, 1954, ch. 2, pp. 161–176 Search PubMed.
- In some cases, the product may precipitate from the reaction media through the reaction progress at room temperature. In these cases, the almost pure product can be separated from the reaction mixture via simple filtration before solvent evaporation.
Footnote |
| † Electronic supplementary information (ESI) available: H NMR and C NMR spectra of the products. See DOI: 10.1039/c6ra17814f |
| This journal is © The Royal Society of Chemistry 2016 |