
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Benzimidazole based Pt(II) complexes with better normal cell viability than cisplatin: synthesis, substitution behavior, cytotoxicity, DNA binding and DFT study†
Ishani
Mitra
a,
Subhajit
Mukherjee
a,
Venkata P.
Reddy B.
a,
Subrata
Dasgupta
a,
Jagadeesh C.
Bose K
b,
Sandip
Mukherjee
c,
Wolfgang
Linert
d and
Sankar Ch.
Moi
*a
aDepartment of Chemistry, National Institute of Technology, Durgapur-713209, W.B., India. E-mail: sankarmoi67@yahoo.com
bDepartment of Bio-Technology, National Institute of Technology, Durgapur-713209, W. B., India
cDepartment of Zoology, Visva-Bharati University, Santiniketan 731235, India
dInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt, 9/163-AC, 1060, Vienna, Austria
First published on 9th August 2016
Abstract
cis-[Pt(ambim)Cl2] 1 (where, ambim = 2-aminomethylbenzimidazole) has been synthesized and characterized by spectroscopic methods. Reaction kinetics between the hydrolyzed product, cis-[Pt(ambim)(H2O)2]2+2 with DL-penicillamine (DL-pen) and glutathione (GSH) have been studied spectrophotometrically in aqueous medium. At pH 4.0, the interactions of 2 with the ligands show two distinct consecutive steps. The association equilibrium constant (KE) for the outer sphere complex formation and rate constants for both the steps have been evaluated. Activation parameters (ΔH‡ and ΔS‡) were evaluated using the Eyring equation and an associative mechanism is proposed for both the reactions. Computational studies using Density Functional Theory (DFT) were carried out to investigate the electronic structures of the complexes. To study the nature of the electronic transitions in complex 1, time dependent DFT was performed. The DNA binding properties of the complexes 2–4 were evaluated by spectroscopic titration, fluorescence indicator displacement experiments and electrophoresis measurements. The complexes effectively bind to calf-thymus DNA via different binding modes with intrinsic binding constants (Kb) in the range of 2.22 × 104 to 4.76 × 104 M−1 which was supported by molecular docking studies. The antiproliferative properties of 2–4 were probed in vitro against human cervical cancer, non-small cell lung carcinoma and hepatocellular liver carcinoma cell lines and 2 was found to be most effective in growth inhibition in all the cell lines. Remarkably, the complexes also generate lower levels of reactive oxygen species (ROS) than cisplatin and have almost no adverse effects on normal cells.
Introduction
Since its arrival in the clinic in 1978, cisplatin1 has played a vital role in cancer chemotherapy, either unaided or in combination with other chemotherapeutics.2 The activity of cisplatin arises from its ability to bind to DNA via covalent cross-links, predominantly by a 1,2 intrastrand crosslink between the [Pt(NH3)2]2+ unit and two adjacent guanine bases of DNA.1 Despite its widespread use, the therapeutic success of cisplatin is marred by several adverse effects, and both inherent and acquired resistances limit its efficacy.3–6 Moreover, recent studies have shown that its exposure in several tissues leads to oxidative damage in vivo, indicating that oxidative stress plays a role in the pathogenesis of cisplatin-induced dose limiting toxicities.7 Unfortunately, its successors, carboplatin and oxaliplatin have not yet demonstrated significant advantages over cisplatin and hence there is an increasingly growing interest in the development of alternative strategies to deliver platinum drugs with enhanced efficacy and cellular uptake and reduced drug resistance.8–15Non-covalent DNA recognition might be a powerful way for metal complexes to exert an anticancer effect through unique molecular actions. Many platinum complexes bearing π-conjugated heterocyclic ligands have been developed and their anti-cancer properties investigated.16–18 The planar aromatic ligand allows these complexes to intercalate non-covalently between the base pairs of DNA. Additionally, inclusion of sterically demanding ligands has also yielded cytotoxic metal complexes which act as DNA groove binders.19–21 With the aim of exploring the ability of new metal complexes as intercalators or DNA groove binders, we have prepared the platinum(II) complex, [Pt(ambim)Cl2], 1, its diaqua analogue, [Pt(ambim)(H2O)2]2+, 2 and the substituted complexes [Pt(ambim)(DL-pen)]+, 3 and [Pt(ambim)(GSH)], 4, where ambim = 2-aminomethylbenzimidazole. The chemistry and pharmacology of benzimidazoles has been of immense relevance to medicinal chemistry as its derivatives possess various biological activities.22–24 Ambim has in fact been utilized to develop innovative compounds for the treatment of cancer.25,26 In this work, cytotoxicity of the carrier ligand ambim and the synthesized Pt(II) complexes were evaluated on different tumor cell lines in comparison to cisplatin. To assess the effect on normal cells upon treatment with the complexes and cisplatin, assays for viability and reactive oxygen species (ROS) generated were also performed. Moreover, DNA binding properties of the complexes have been reported along with docking studies to rationalize the interactions.
DNA platination is commonly accepted as the ultimate event in the mechanism of action of platinum anticancer drugs.27 However, already in the blood, where the Pt drug is administered by injection or infusion, several sulfur-donor ligands are available for kinetic and thermodynamic competition.28 It is hypothesized that the platinum temporarily binds to the S-containing bio-molecules in the cell, forming an intermediate which acts as a platinum reservoir,28–30 before finally interacting with guanine-N7 of DNA. The Pt–S interactions are believed to be kinetically preferred but the Pt–DNA adducts are thermodynamically more stable under physiological conditions. In this paper, kinetic measurements were carried out to study the substitution reactions of complex 2 with biologically relevant nucleophiles. DL-Penicillamine (β,β′-dimethyl cysteine), is a degradation product of β-lactum antibiotic, penicillin, and a S-containing bioactive ligand, while glutathione (g-Glu-CysH-Gly, GSH) is the most prevalent intracellular thiol with concentrations up to 10 mM and is the most abundant low molecular weight peptide. The study of the chemical transformations of cytotoxic Pt(II) complexes with sulfur containing biologically relevant nucleophiles provides a fundamental basis for understanding toxicity and development of resistance in biological systems and hence, is of special concern for pharmaceutical and biomedical research.
Experimental
Reagents
All chemicals used for the kinetic and bio-activity study were of highest purity as available. The starting compounds K2PtCl4 (99%), 2-aminomethylbenzimidazole (ambim) (98%), DL-penicillamine (97%) and glutathione (98%) were purchased from Sigma Aldrich. AgClO4 (97%), AgNO3 (99%), K2HPO4 and NaClO4 were purchased from Merck. All these reagents were used without further purification. For kinetic purpose, double distilled water was used to prepare all kinds of solutions and molecular biology grade water was used for bioactivity study. All solvents used were of analytical grade. Cisplatin (Cis-gland) was purchased from Gland Pharma Limited. Dulbecco's modified Eagle medium (DMEM), phosphate-buffered saline (PBS) and fetal bovine serum (FBS) were purchased from Hi-Media. Agarose, nitrotetrazolium blue chloride (NBT), Tris-borate-EDTA (TBE) and ethidium bromide (EtBr) were purchased from Sigma-Aldrich and used in gel electrophoresis experiment.Physical measurements
Elemental analysis of the complexes was carried out on a Perkin-Elmer 2400 series-II CHNS/O Analyser. The pH measurements were done with the help of a Eutech digital pH meter (pH Tutor) with accuracy of ±0.01 U, calibrated with a standard phosphate buffer solution (KH2PO4/Na2HPO4). Fourier transform infrared (FTIR) spectra of the complexes were recorded on a Nicolet-iS-10 spectrometer (KBr disk, in the range of 4000–400 cm−1). The kinetic measurements and absorption titrations were performed with a Shimadzu UV 1800 spectrophotometer attached to a thermoelectric cell temperature controller (Model TCC-100, accuracy ±0.1 °C). Emission spectra were recorded using a HITACHI F-2500 Fluorescence spectrophotometer. 1H NMR spectra were recorded with a Bruker Avance-400 (400 MHz) spectrometer using D2O or DMSO-d6 as solvent. 13C NMR was recorded on a Bruker Avance-200 (200 MHz). Waters Q-TOF Micro YA263 Mass spectrometer was used to record ESI-MS in water. The pKa values of the complex 2 were determined by Metrohm 888 Titrando titro-processor. Stat Fax™® 2100 Microplate (USA) Enzyme-linked immunosorbent assay (ELISA) plate reader was used for MTT assay. The agarose gel electrophoresis was carried out in Bio-Rad Mini-Sub Cell GT System. The gel picture was taken with Imager Gel Doc™ EZ. Ziess Axio Observer. Z1 inverted microscope was used to capture the cell images with 100× magnification.Syntheses of complexes
Elemental analysis of complex 1 (C8H9N3Cl2Pt) gave; C = 23.10% (23.24); H = 2.38% (2.20) and N = 9.84% (10.17) where the numbers in parentheses were calculated from the chemical formula.
Electronic absorption spectrum of 1 in DMF, (λmax/nm) (ε/M−1 cm−1): 282 (5757). Selected IR frequencies (KBr disk, cm−1): 3570(m), 3488(br), 3235–3200(br), 3132(m), 3081(ms), 2964(m), 2927(w), 2788(br), 1620(s), 1578(s), 1477(s), 1454(s), 1417(s), 1385(s), 1284(s), 1154(br), 1002(s) and 740(s) (ESI Fig. S1†). 1H NMR (200 MHz, DMSO-d6) of complex 1 (δ in ppm and J in Hz): δ 13.79 (s, 1Hd), δ 8.54 (d, J = 6.0, 1Hk) δ 7.58 (d, J = 8.0, 1Hf), δ 7.46–7.20 (m, 2Hg,h), δ 6.12 (brt, 2Ha), δ 4.04 (t, J = 6.0, 2Hb) and δ 3.38 (water in DMSO) (ESI Fig. S2†). 13C NMR (200 MHz, DMSO-d6) of complex 1 (δ in ppm) (ESI Fig. S3†): Cc (162.75), Cl (139.30), Ce (138.45), Cg (123.64), Ch (122.71), Ck (117.59), Cf (117.38), Cb (39.46, m).
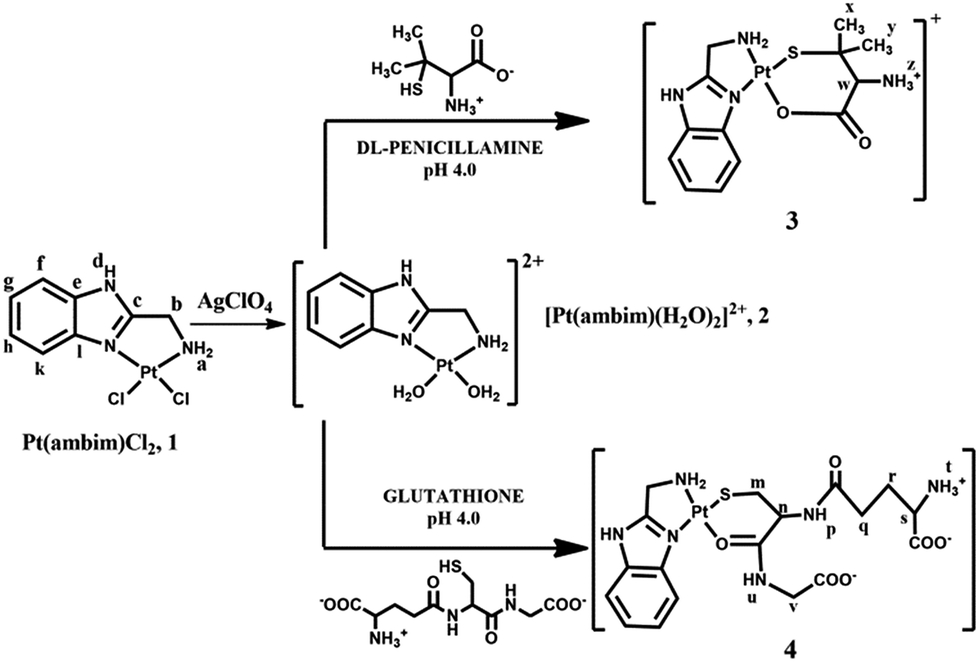 | ||
| Fig. 1 Structures of complexes and ligands. | ||
Elemental analysis of complex 2 (C8H13N3O10Cl2Pt) gave; C = 16.24% (16.65); H = 2.22% (2.27) and N = 7.35% (7.28) where the numbers in parentheses were calculated from the chemical formula. Electronic absorption spectrum of 2 in water, (λmax/nm) (ε/M−1 cm−1): 271 (5946). Selected IR frequencies (KBr disk, cm−1): 3629(m), 3424(br), 3130(m), 2923(w), 1636(m), 1596(m), 1559(m), 1545(m), 1477(s), 1457(m), 1401(s), 1282(m), 1085(s), 939(m), 745(m), 625(s) and 443(m) (ESI Fig. S4†). 1H NMR (400 MHz, DMSO-d6) of complex 2 (δ in ppm and J in Hz): δ 8.03 (d, J = 6.4, 1Hk) δ 7.92 (d, J = 3.6, 1Hf), δ 7.41–7.38 (m, 2Hg,h), δ 6.32 (brs, 2Ha), δ 4.03 (s, 2Hb), δ 3.69 (coordinated water) and δ 2.50 (solvent peak) (ESI Fig. S5†).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand (M:L) ratio for the reaction between complex 2 with the ligands at pH 4.0. Both were mixed according to their probable ratios, like 1:1, 1:2, 2:1 etc. and heated to near about 50 °C for few hours and absorbances were measured till a constant maximum was reached. A bell shaped curve was obtained for both DL-pen and GSH when absorbance vs. [M]/[M] + [L] was plotted (ESI Fig. S6 and S7†) and the M:L ratio was found to be 1:1 which indicates that both act as bidentate ligands. The development of a characteristic peak in the product complexes 3 and 4 at 265 and 273 nm respectively was monitored as a function of time at different fixed temperatures (ESI Fig. S8 and S9†). Complex 2 and the ligands were mixed in 1:1 molar ratio at pH 4.0 in accordance with Job's result to obtain solid products by slow evaporation.
:ligand (M:L) ratio for the reaction between complex 2 with the ligands at pH 4.0. Both were mixed according to their probable ratios, like 1:1, 1:2, 2:1 etc. and heated to near about 50 °C for few hours and absorbances were measured till a constant maximum was reached. A bell shaped curve was obtained for both DL-pen and GSH when absorbance vs. [M]/[M] + [L] was plotted (ESI Fig. S6 and S7†) and the M:L ratio was found to be 1:1 which indicates that both act as bidentate ligands. The development of a characteristic peak in the product complexes 3 and 4 at 265 and 273 nm respectively was monitored as a function of time at different fixed temperatures (ESI Fig. S8 and S9†). Complex 2 and the ligands were mixed in 1:1 molar ratio at pH 4.0 in accordance with Job's result to obtain solid products by slow evaporation.
Elemental analysis of complex 3 (C13H19N4O6ClSPt) gave; C = 26.22% (26.47); H = 3.21% (3.25) and N = 9.14% (9.50) where the numbers in parentheses were calculated from the chemical formula. Electronic absorption spectrum of 3 in water, λmax/nm (ε/M−1 cm−1): 265 (3552). Selected IR frequencies (KBr disk, cm−1): 3448(br), 3121(sh), 2967(w), 2925(w), 1640(s), 1458(m), 1345(m), 1088(s), 940(w), 741(m), 626(s), and 473(s) (ESI Fig. S10†). 1H NMR (400 MHz, δ in ppm DMSO-d6) data for complex 3: δ 8.51 (d, J = 6.4 Hz, 1Hk), δ 8.44 (d, J = 6.4 Hz, 1Hf), δ 8.27–8.16 (m, 2Hg,h), δ 6.56 (brt, 2Ha), δ 6.10 (brs, 3Hz), δ 4.09 (d, J = 6.0 Hz, 2Hb), δ 3.48 (s, 1Hw), δ 3.44 (water in DMSO), δ 2.53 (solvent peak), δ 2.35 (s, 3Hx) and δ 2.19 (s, 3Hy) (ESI Fig. S11†). ESI-MS of 3 in water: m/z 131.04, 146.06, and 490.13 (ESI Fig. S12†).
Elemental analysis of complex 4 (C18H24N6O6SPt) gave; C = 33.17% (33.38); H = 3.38% (3.74) and N = 12.64% (12.98) where the numbers in parentheses were calculated from the chemical formula. Electronic absorption spectrum of 4 in water, λmax/nm (ε/M−1 cm−1): 273 (3053). Selected IR frequencies (KBr disk, cm−1): 3420(br), 3267(br), 3026(br), 2923(w), 1644(s), 1537(s), 1396(s), 1277(m), 1088(s), 748(m), 626(s) and 436(w) (ESI Fig. S13†). 1H NMR (400 MHz, δ in ppm, D2O) data: δ 7.87 (d, J = 3.6 Hz, 1Hk), δ 7.69 (d, J = 2 Hz, 1Hf), δ 7.44–7.42 (m, 2Hg,h), δ 6.46 (s, 2Ha), δ 4.58 (s, 1Hn), δ 4.19 (s, 2Hb), δ 3.87 (s, 1Hv), δ 3.76 (t, J = 5.2 Hz, 1Hs), δ 2.96 (m, 2Hm), δ 2.49–2.52 (m, 2Hq) and δ 2.12–2.15 (m, 2Hr) (ESI Fig. S14†). ESI-MS of 4 in water: m/z 131.04, 146.06, 454.08 and 646.04 (ESI Fig. S15†).
Synthesis safety note: perchlorate salts of metal complex should be handled cautiously as these are hazardous and explosive upon heating. No explosive event was encountered in the present study.
Determination of pKa values of complex 2
The pKa values of 2 were determined by potentiometric measurement using standard 0.025 M KOH solution. Standard buffer solutions prepared according to NBS specifications were used to calibrate the electrode and titro-processor. pKa values were measured by pH versus volume titration of 1.50 × 10−4 (M) complex 2 and the ionic strength was adjusted to 0.1 M with NaClO4, at 25 °C. The pH was plotted against volume of base consumed (Fig. 2).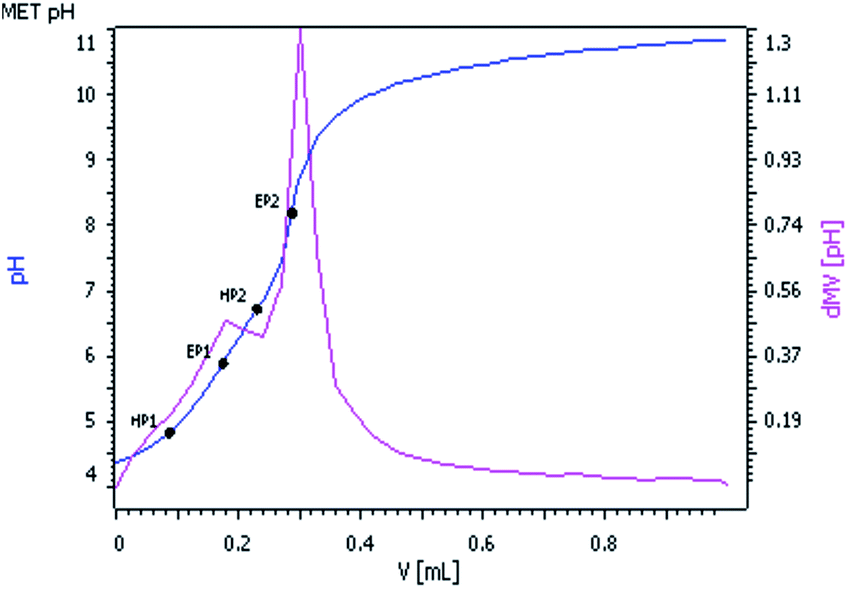 | ||
| Fig. 2 pH vs. volume (mL) of 0.025 M KOH solution. | ||
Two equivalent points EP1 and EP2 were observed in the titration curve corresponding to the pKa values as HP1 and HP2. The pKa1 and pKa2 values of complex 2 were found to be 4.98 and 6.65 respectively, which are comparable with the reported pKa values of similar [Pd(ambim)(H2O)2]2+ system.32
Kinetic investigation
The usual mixing technique was followed and pseudo-first-order reaction conditions were employed throughout the kinetic runs for the reactions. The progress of the reaction was followed by measuring the increase in absorbance for the ligands with complex 2 at 240 nm and 245 nm respectively, where the spectral differences between complex 2 and the product complex 3 and 4 is at a maximum. The pH and ionic strength were maintained at 4.0 and 0.1 mol dm−3 (with NaClO4) respectively. The method of Weyh and Hamm33 was adopted to calculate the rate constants for both the kinetic studies. The rate data, represented as an average of duplicate runs, were reproducible to within ±4%.Computational details
All geometry optimization and energy calculations of complex 1–4 were performed using Density Functional Theory (DFT) at the B3LYP34,35 level of theory for the complexes 1–4 using Gaussian 09 software.36 Global minimum optimized structures were confirmed by frequency analysis at the same level of theory. The relativistic effective core potential (ECP) and associated valence double ξ (zeta) basis set of Hay and Wadt37,38 (LANL2DZ) were employed for Pt atom. The standard split valence basis set 6-31G(d)39,40 was applied for carbon, oxygen, nitrogen, hydrogen and sulfur. Natural bond orbital41 (NBO) analysis was performed to get an insight into the electronic structure and bonding of complexes 3 and 4. To evaluate the nature of the orbitals involved in the transitions related to the experimental observations, Time Dependent-Density Functional Theory (TD-DFT) computational method was applied.Molecular docking studies were performed using the AutoDock Vina program.42 Two structures of B-DNA were selected: a dodecamer d(CGCGATATCGCG)2 (1DNE) for groove binding and an octamer d(GAAGCTTC)2 (1DSC) for intercalation.43 Each of the complexes was docked with B-DNA. The water molecules and the ligands were removed from the B-DNA before carrying out docking calculations. The binding site was centred on DNA and a grid box was generated with 40 × 40 × 60 points and a 1.0 Å grid spacing involving almost entire macromolecule. All other parameters were kept at their default values.
DNA binding studies
All spectroscopic titrations were carried out at room temperature in Tris–HCl buffer (5 mM Tris–HCl, 50 mM NaCl, pH 7.4, Tris = Tris(hydroxymethyl)amino methane).Interaction of complexes 2–4 with CT-DNA has been studied with UV spectroscopy in order to investigate the probable binding mode. The absorption titration experiments were performed with a constant concentration of the complexes (20 μM), while varying [complex]/[CT DNA] mixing ratio in the range 0.1 to 1.0. To obtain the absorption spectra, the required amount of CT-DNA was added to both sample and reference cell to eliminate the absorbance of CT-DNA itself. Complex-DNA solutions were allowed to incubate for 15 minutes before the absorption spectra were recorded. From the absorption titration data, the binding constant (Kb) was determined using the equation44
| [DNA]/(εa − εf) = [DNA]/(εb − εf) + 1/{Kb(εb − εf)} |
The molecular fluorophore EtBr (3,8-diamino-5-ethyl-6-phenylphenanthridinium bromide) emits intense fluorescence in the presence of CT-DNA due to its strong intercalation between the adjacent DNA base pairs. The study involves the addition of the complexes to DNA pretreated with EtBr and the measurement of the intensity of emission. The DNA–EtBr complex was prepared by adding 20 μM EtBr and 20 μM CT-DNA in buffer. The interaction of complexes 2–4 with the DNA–EtBr complex was studied by adding a certain amount of the complex solution step by step into the solution of DNA–EtBr complex. The effect of the addition was obtained by recording the variation of fluorescence emission spectra with excitation wavelength at 537 nm and emission wavelength at 550 to 750 nm after 15 minutes of incubation period in each case. The complexes under study do not show any fluorescence emission band at room temperature in solution or in the presence of CT-DNA under the same experimental conditions. Therefore, the observed quenching is characteristic to the displacement of EtBr from its EtBr–DNA complex. To investigate the quenching efficiency, the spectra were analyzed according to the classical Stern–Volmer equation45
| I0/I = 1 + KSV[Q] |
| KEtBr[EtBr] = Kapp[Q] |
| log(F0 – F)/F = logKF + nlog[Q] |
The plot of log(F0 − F)/F versus log[Q] is drawn and fitted linearly, and then the n value, the number of binding sites per nucleotide, can be obtained from the slope.
Bioactivity studies
Normal healthy Swiss albino mice were anesthetised and then liver and lungs tissues were removed after perfusion with sterile PBS. These tissues were minced and digested in PBS containing 0.5% trypsin for 45 minutes at 37 °C. Cells were separated after tissue digestion by filtration through nylon mesh followed by several wash with PBS. These cells were then kept in DMEM containing 2% FBS and antibiotics, and finally resuspended in the medium and plated in a six well plate, incubated in a humidified 95% O2/5% CO2 atmosphere at 37 °C.
The colorimetric MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyl tetrazolium bromide) assay48,49 was employed to determine the cytotoxicity of the compounds under study. Approximately 10000–12000 cells were added to each well of a 96-well plate in DMEM medium. Different doses of ambim, complex 2–4 and cisplatin ranging from 5.0 to 50.0 μM concentrations were applied in triplicates for 48 h. Untreated cells and cells treated with cisplatin were used as negative and positive control50 respectively. 20 μL MTT (5 mg mL−1 in PBS) solution was added to each well and incubated for another 3 h. 150 μL of DMSO was added to each well to dissolve the blue formazan product. The absorbance of this product was measured at 540 nm, using ELISA plate reader.51 The IC50 (the concentration of tested agent that caused 50% inhibition of cell growth) values were obtained from the results of triplicate determinations of at least three independent experiments. Similar procedure was followed for the evaluation of viability of mice liver and lung cells.
Results and discussion
Spectroscopic analyses
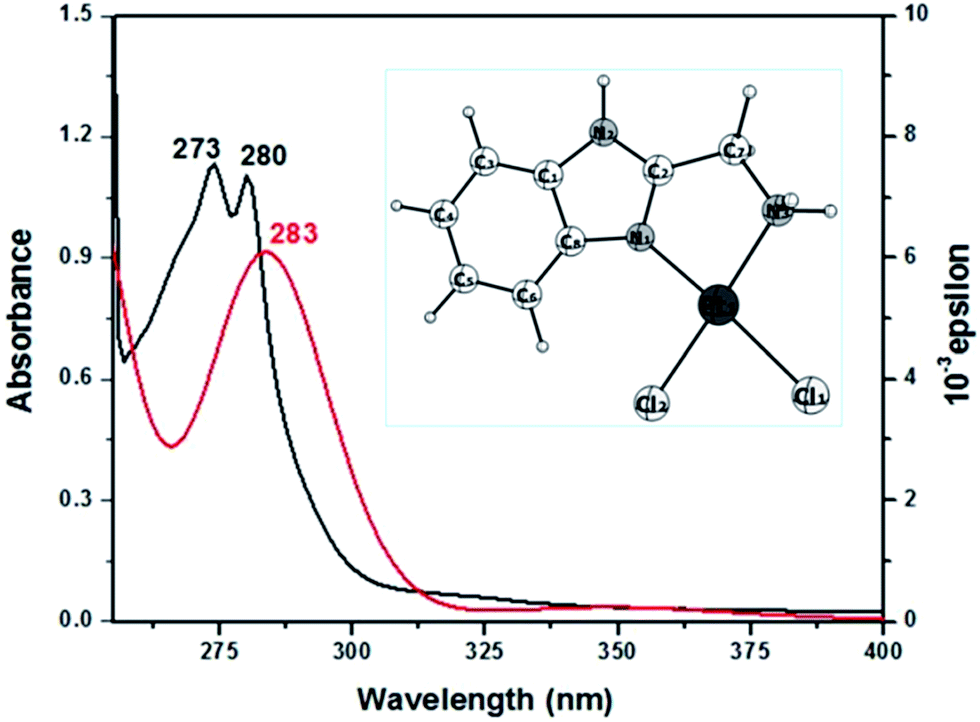 | ||
| Fig. 3 UV-vis (in black) and simulated TD-DFT (in red) spectrum of complex 1 in DMSO. Inset: optimised structure of complex 1. | ||
| Experimental band (ε dm3 mol−1 cm−1) | TD-DFT peaks (oscillator strength) | Major contribution |
|---|---|---|
| 280 | 283 (0.0715) | HOMO → L+1 (95%) |
| 273 | 274 (0.0038) | H−1 → L+1 (91%) |
Electronic spectra of the substituted products 3 and 4 display different λmax values in aqueous medium with hypsochromic (∼6 nm) and bathochromic (∼2 nm) shift with respect to complex 2. The spectral changes of the products are due to n → π* and π → π* transitions of the coordinated ligands in complex 3 and 4, which indicate the formation of new products.
The peak appearing at 3026 cm−1 for the zwitterionic form of GSH56 due to N–H stretching in a H-bonded NH3+, shows no considerable shift in the spectrum of complex 4. The strong band at 2525 cm−1 due to ν(S–H) is also absent indicating coordination by thiol group and a band at 436 cm−1 can be assigned to ν(Pt–S). The bands at 1537 and 1396 cm−1 in GSH are assigned to νas(COO−) and νs(COO−)57 which show no considerable shift in the spectrum of 4 suggesting non-participation of the carboxylate group in complex formation. Moreover, the band at 1713 cm−1 is due to the ![[double bond splayed left]](https://www.rsc.org/images/entities/char_e009.gif) C
C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of the carboxylate group of glycine residue of GSH, which confirms its non-involvement in coordination to Pt(II).56 The band at 1665 cm−1 in free GSH assigned to CO stretching of peptide bond, has shifted to lower frequency (1644 cm−1) indicating coordination of carbonyl group of the cysteine residue to Pt(II)58 resulting in a preferred six-membered chelate.
O stretching vibration of the carboxylate group of glycine residue of GSH, which confirms its non-involvement in coordination to Pt(II).56 The band at 1665 cm−1 in free GSH assigned to CO stretching of peptide bond, has shifted to lower frequency (1644 cm−1) indicating coordination of carbonyl group of the cysteine residue to Pt(II)58 resulting in a preferred six-membered chelate.
A decrease in pH on mixing complex 2 with DL-pen (from 4.0 to 2.71) and GSH (from 4.0 to 2.96) corroborates with sulphur coordination. The high affinity of Pt(II) for sulphur provides the driving force for deprotonation of the thiol groups in the first step of complex formation. These findings are consistent with bidentate coordination through S and O atoms shown by DL-pen and GSH in complexes 3 and 4.
The resonance signals of the α and β protons of cysteine residue in the GSH moiety appear at slightly downfield positions in the spectrum of 4, which corroborates the coordination of Pt(II) by thiol and peptide carbonyl. The acidic protons are missing in the spectrum due to exchange with the solvent D2O. The protons in the aromatic region appear at 8.51–8.27 ppm and 7.87–7.44 ppm in complexes 3 and 4 respectively (as compared to 8.03–7.38 ppm in complex 2), confirming the presence of ambim moiety in the complexes.
ESI mass
The formation of metal complexes and various ionic forms in aqueous solution were studied with ESI-MS. The peak at m/z 490 in the spectrum of 3 indicates the formation of the product [Pt(II) + ambim + DL-pen-H+]+. Similarly for complex 4, molecular ion peak corresponding to [Pt(II) + ambim + GSH-2H+] is found at m/z 646. The molecular ion peaks of 3 and 4 were accompanied by isotope peaks with expected relative abundance. Base peak obtained at m/z 131 for both the complexes can be tentatively assigned to a methylium cation generated by the loss of –NH2 unit from ambim. A prominent peak at m/z 146 observed in the spectra of 3 and 4 can be attributed to just the ambim moiety after removal of platinum metal and the ligand. The expulsion of ambim and –CO2 from the GSH moiety, probably gives rise to the peak at m/z 454 in the spectrum of 4.Kinetic study
The acid dissociation constants of DL-pen and GSH at 25 °C are 1.81(–COOH) 7.96(NH3+), 10.72(SH) and 2.12(COOH), 3.59(COOH), 8.75(NH3+), 9.65(S–H) respectively.60,61 Thus, both DL-pen and GSH exist in their zwitterionic forms at pH 4.0. The pKa1 and pKa2 values of the complex 2 were found to be 4.98 and 6.65 respectively. We can thus assume that at pH 4.0 the reactant complex 2 exists as diaqua species. At a particular temperature, pH 4.0 and fixed concentration of complex 2, the plots of ln(Aα − At) versus time (where, Aα and At are the absorbances at infinite time i.e. after the completion of the reaction and at time t) for various ligand concentrations were curved at the initial stage and subsequently of constant slope (Fig. 4 for DL-pen and ESI Fig. S17† for GSH). This indicates that the reaction proceeds through a two-step consecutive process and we propose that in the first step one aqua ligand is replaced from 2 by the sulphur atom of the ligands. The second step is slower, where the other aqua molecule is substituted and ring closure takes place.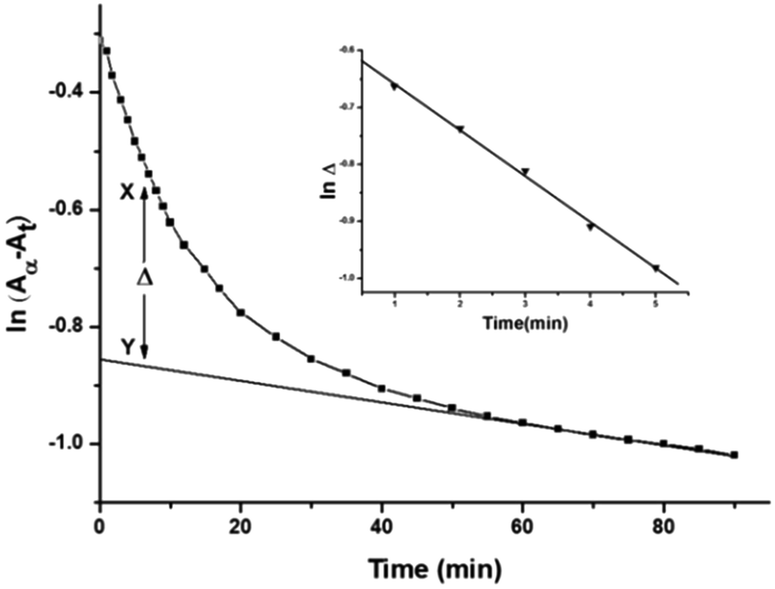 | ||
| Fig. 4 A typical plot of ln(Aα − At) vs. time: [complex 2] = 1.50 × 10−4 M; [DL-pen] = 1.50 × 10−3 M; 25 °C. Inset: typical plot of lnΔ vs. time (min). | ||
The rate constants for such a two step process can be evaluated by assuming the following scheme (Scheme 1):
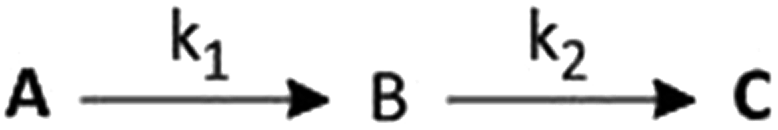 | ||
| Scheme 1 | ||
Where, A is the diaqua complex 2, B is the sulfur bonded (one aqua ligand substituted intermediate, and C is the final product, 3 and 4. Formation of C from B is predominant after some time has elapsed.
Evaluation of k1 for A → B step
At each temperature, the rate constant k1(obs) values were evaluated by the method of Weyh and Hamm33 from the plots of lnΔ (the variation of Δ is shown in Fig. 4, inset) versus time (where t is small) and are collected in ESI Table S1.† Using the usual consecutive rate law:| (Aα − At) = a1exp(−k1(obs)t) + a2exp(−k2(obs)t) | (1) |
exp(−k2(obs)t)] were obtained from the parameter X–Y at different times.| lnΔ = constant − k1(obs)t | (2) |
Δ versus t.
The same procedure was followed in the range of ligand concentration from 1.50 × 10−3 mol dm−3 to 4.50 × 10−3 mol dm−3, at constant complex 2 concentration (1.50 × 10−4 mol dm−3) and at different temperatures (25, 30, 35, 40 and 45 °C). The reaction rate increases with increasing ligand concentration up to a limiting value (ESI Fig. S18† for DL-pen and S19 for GSH). The dependency of the k1(obs) values on ligand concentration can be explained in terms of rapid formation of an outer sphere association complex between the reactant complex 2 and ligand in the step A → B. Since the metal ion reacts with its immediate surroundings, further change in ligand concentration beyond the saturation point will not affect the reaction rate and a gradual approach towards limiting rate is observed. This is followed by inner sphere association through sulfur coordination via displacement of one water molecule resulting in the intermediate B (Scheme 2). The following scheme can be proposed for the reaction:
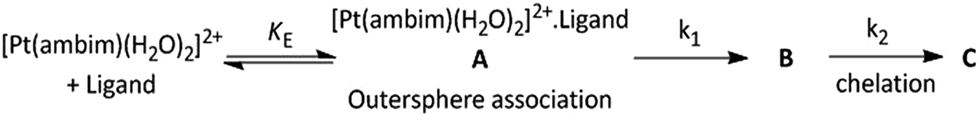 | ||
| Scheme 2 | ||
Based on the above equations, a rate expression (6) can be derived for the A → B step:
| d[B]/dt = k1KE[2][ligand]/(1 + KE[ligand]) | (3) |
| d[B]/dt = k1(obs)[2]T | (4) |
| 1/k1(obs) = 1/k1 + 1/k1KE[ligand] | (5) |
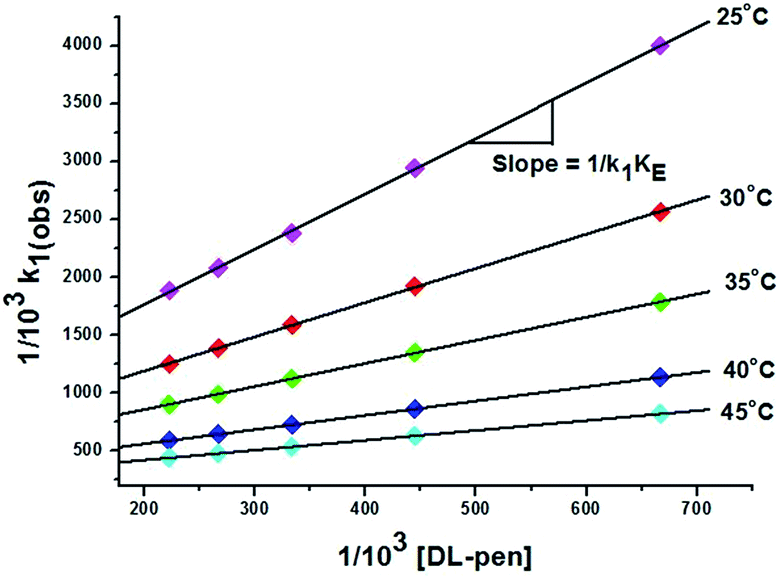 | ||
| Fig. 5 Plot of 1/103 × k1(obs)versus 1/103 [DL-pen] at different temperatures. | ||
| Temp. (°C) | DL-pen | GSH | ||||
|---|---|---|---|---|---|---|
| 103k1 (M−1 s−1) | K E (dm3 mol−1) | 105k2 (s−1) | 103k1 (M−1 s−1) | K E (dm3 mol−1) | 105k2 (s−1) | |
| 25 | 1.20 ± 0.10 | 176.41 ± 0.21 | 1.17 ± 0.01 | 3.47 ± 0.08 | 360.57 ± 0.12 | 3.15 ± 0.05 |
| 30 | 1.65 ± 0.15 | 206.65 ± 0.16 | 1.46 ± 0.04 | 4.20 ± 0.12 | 372.91 ± 0.17 | 4.00 ± 0.03 |
| 35 | 2.18 ± 0.09 | 229.16 ± 0.19 | 1.98 ± 0.03 | 5.29 ± 0.10 | 406.39 ± 0.22 | 4.82 ± 0.04 |
| 40 | 3.16 ± 0.11 | 258.32 ± 0.24 | 2.61 ± 0.05 | 6.40 ± 0.09 | 448.82 ± 0.18 | 5.76 ± 0.02 |
| 45 | 4.02 ± 0.07 | 289.44 ± 0.14 | 3.44 ± 0.01 | 8.06 ± 0.14 | 490.44 ± 0.13 | 6.87 ± 0.03 |
Evaluation of k2 for B → C step
The second step involves displacement of another water molecule accompanied by ring closure through the second donor site of the ligands. Due to steric hindrance, this chelation step is slower and independent of ligand concentration. At each temperature, the k2(obs) values were calculated from the limiting linear portion (when t is large) of the plot of ln(Aα − At) versus t (Fig. 4) and the values are collected in ESI Table S2.† Unlike k1, k2 values (listed in Table 2) were found to be independent of ligand concentration at all temperatures studied.The derived rate equation of the reactions based on kinetic observations is as follows,
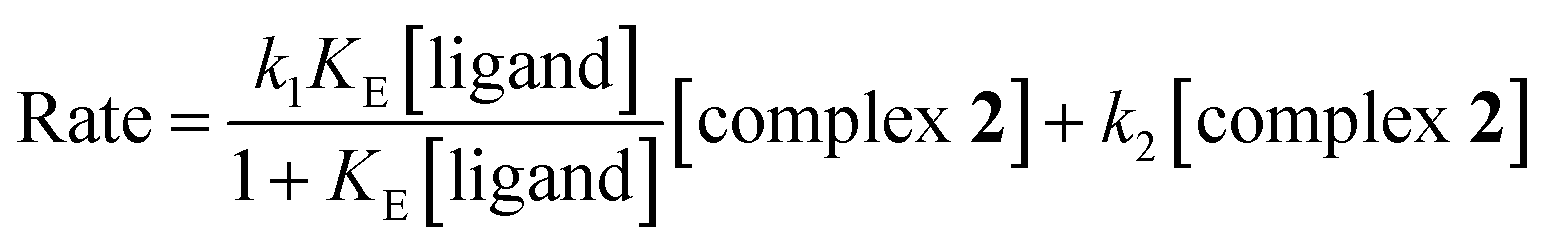 | (6) |
Effect of temperature and pH
The reactions were studied at five different temperatures at different ligand concentrations and the substitution rate constants for both the steps (k1 and k2) are given in Table 2. The activation parameters calculated from Eyring plots (ESI Fig. S21 and S22† for DL-pen and S23, S24 for GSH) are given in Table 3 and compared with those for substitution reactions on analogous square planar Pt(II) systems (ESI Table S3†).| Systems | ΔH‡1 (kJ mol−1) | ΔS‡1 (J K mol−1) | ΔH‡2 (kJ mol−1) | ΔS‡2 (J K mol−1) |
|---|---|---|---|---|
| [Pt(ambim)(H2O)2]2+/DL-pen | 43.79 ± 1.31 | −149.00 ± 1.20 | 38.7 ± 1.16 | −205.12 ± 1.78 |
| [Pt(ambim)(H2O)2]2+/GSH | 31.04 ± 1.44 | −184.65 ± 1.17 | 26.46 ± 1.19 | −239.16 ± 1.61 |
The reactions were studied at six different pH values. At a fixed [complex 2], [ligand] and 0.1 M ionic strength, k(obs) values for both the steps increases initially from pH 3.0 to 4.0 and decreases from 5.0 to 7.4. For formation of complex 3, 103 × k1(obs) values at 25 °C were 0.19, 0.25, 0.17, 0.12, 0.09 and 0.05 s−1 and 105 × k2(obs) values were 0.94, 1.03, 0.86, 0.74, 0.52 and 0.41 s−1 at pH 3.0, 4.0, 5.0, 6.0, 6.8 and 7.4 respectively. Similar trend is observed in case of complex 4. The increase in rate may be explained on the basis of acid dissociation equilibria of the ligands and complex 2. In the studied pH range, 2 exists as diaqua species till its first pKa1 value (4.98). After that the diaqua species is transformed to a bridging dimer,62,63 which reacts with the ligands at a comparatively slower rate. The rate enhancement up to pH 4.0 might be explained by the deprotonation and increased donor ability of the ligands. At higher pH, the percentage of bridged dimer is maximum, consequently the rate decreases. Hence, the substitution reactions were followed at a constant pH of 4.0 to avoid complication from an additional parameter of [H+] in the rate equation.
Mechanism
Based on the result of Job's method of continuous variation and the spectroscopic data, bidentate ligation by DL-pen and GSH has been suggested. Being a soft base, the sulphur donor centres of the ligands attack the soft Pt(II) centre first. From the low values of enthalpy of activation (ΔH‡1 and ΔH‡2) and high negative values of entropy of activation (ΔS‡1 and ΔS‡2), an associative mechanism is proposed (Scheme 3) for both the ligand substitution reactions. Furthermore, ΔS‡2 is found to be more negative than ΔS‡1, which suggests that compactness has already been achieved in B and the transformation of B to C is by displacement of another aqua ligand through chelation. Thus, complex formation occurs via a rapid outer sphere association, followed by two consecutive steps: the first being dependent of [ligand] and the second being a slower ring closure step which is independent of [ligand]. The plausible mechanism for the interaction of complex 2 with DL-pen and GSH is shown as follows: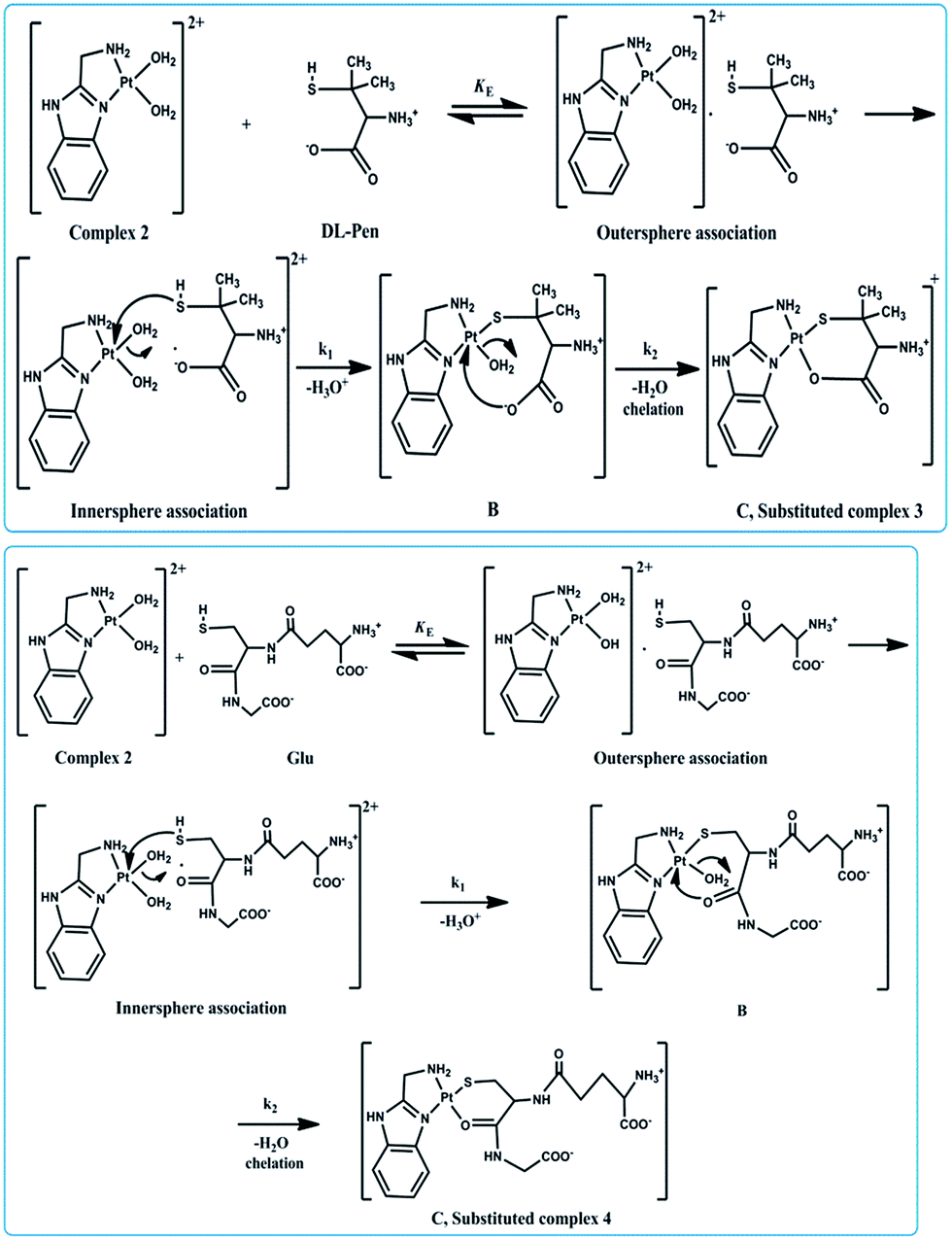 | ||
| Scheme 3 Plausible mechanism of the reactions between complex 2 with DL-pen and GSH. | ||
Computational study
A list of bond distances [Å] and angles [deg] obtained from optimized geometry (ESI Fig. S25†) for complexes 2–4 (comparable with reported data for closely related structures64,65) is presented in Table 4.| 2 | 3 | 4 | References64,65 | |
|---|---|---|---|---|
| Bond distance (Å) | ||||
| Pt–N1 | 1.988 | 2.043 | 2.048 | 2.030 |
| Pt–N3 | 2.068 | 2.179 | 2.196 | 2.039 |
| Pt–S1 | N.A. | 2.327 | 2.326 | 2.356 |
| Pt–O2 | N.A. | 2.040 | 2.037 | 2.055 |
|
||||
| Bond angle (°) | ||||
| N1–Pt–N3 | 80.30 | 79.27 | 79.16 | N.A. |
| N3–Pt–O2 | 100.40 | N.A. | N.A. | N.A. |
| N1–Pt–O1 | 95.94 | N.A. | N.A. | N.A. |
| N3–Pt–O2 | N.A. | 87.88 | 88.40 | N.A. |
| O2–Pt–S1 | N.A. | 95.56 | 94.64 | N.A. |
The surface plots of HOMO and LUMO for complexes 2–4 and their energies are shown in Fig. 6. Positive and negative regions are shown in red and green colours respectively. HOMO and LUMO calculation of complex 2 reveals that electron density of HOMO is localized mostly on benzimidazole ring of carrier ligand ambim and minimum on the metal ion without significant contributions. But the charge density of HOMO for 3 and 4 is localised on DL-pen and glutathione part of the complex respectively, whereas LUMO is localised on ambim.
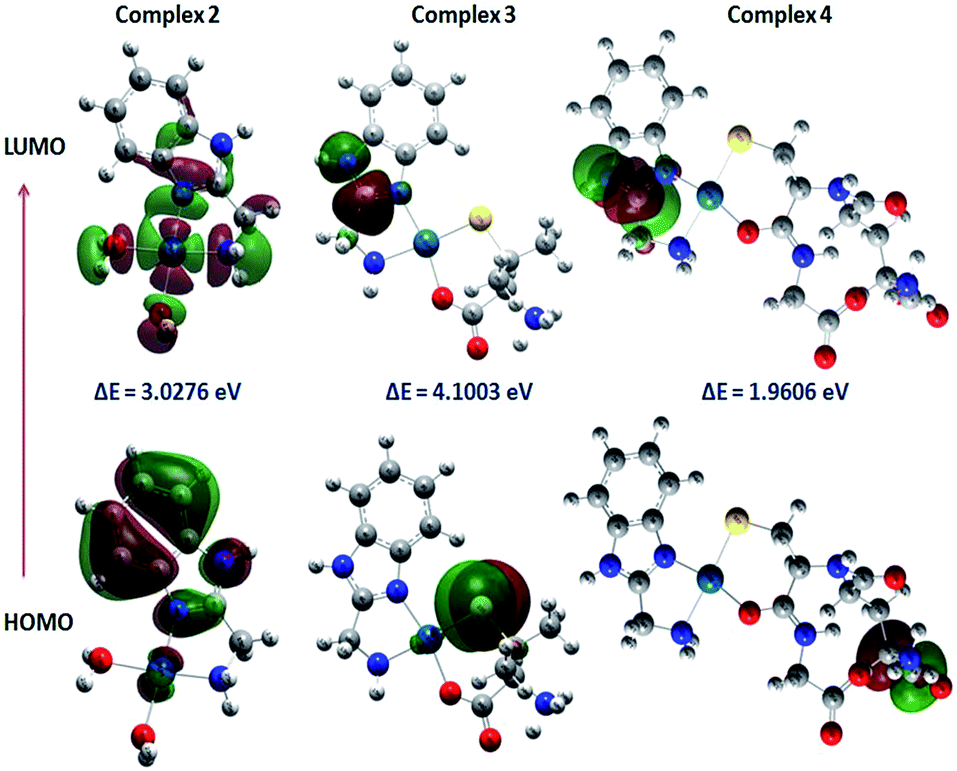 | ||
| Fig. 6 Counter plots of HOMO and LUMO of complexes 2–4. | ||
NBO calculation
NBO analysis provides detailed insight into the nature of electronic structure and bonding in the product complexes 3 and 4 by considering chelation through (S,O) and (S,N) in the substitution reaction. Calculated atomic charges and electronic configurations for complex 3 (S,O) and 4 (S,O) and the probable complexes 3′ (S,N) and 4′ (S,N) (ESI Fig. S26†) are listed in ESI Tables S4 and S5.† By comparing the zero-point corrected energies, it can be concluded that (S, O) coordination is preferred over (S, N), which corroborates the spectroscopic results.Table 5 lists the calculated occupancies (electron densities) on NBO orbitals, hybrids on atoms and polarities of bonds between atoms. According to these results, the platinum atom in 3 and 4 forms sigma bond with S1 atoms of the thiol groups of DL-pen and GSH respectively. The coordination bonds between Pt and the imidazole N1, amino N2 and oxygen O2 (of carboxylate and amidic carbonyl oxygen) can be described as a donation of electron density from donor orbitals to Pt acceptor orbitals.
| Complex | NBO orbitalb | Occupancy (e) | Polarityc (%) | NBO hybrid | AOd (%) |
|---|---|---|---|---|---|
| a Calculations performed at the B3LYP/6-31G(D)/LANL2DZ of theory. b LP(N/O) is a valence lone pair orbital on nitrogen/oxygen atom. c Values for the A–B sigma molecular orbital. d Percentage contribution of atomic orbitals in NBO hybrid. | |||||
| 3 | σ(Pt–S1) | 1.9797 | 38.05(Pt) | sd3.03 | s(23.13%)p(6.86%)d(70.01%) |
| 61.95(S1) | sp6.26 | s(13.74%)p(86.03%)d(0.23%) | |||
| LP(O2) | 1.9249 | — | sp1.66 | s(37.60%)p(62.36%)d(0.04%) | |
| LP(N1) | 1.6191 | — | sp2.79 | s(26.38%)p(73.61%)d(0.00%) | |
| LP(N3) | 1.7256 | — | sp4.92 | s(16.90%)p(83.10%)d(0.00%) | |
| 4 | σ(Pt–S1) | 1.9823 | 39.73(Pt) | sd3.10 | s(22.84%)p(6.38%)d(70.78%) |
| 60.27(S1) | sp6.68 | s(12.99%)p(86.77%)d(0.24%) | |||
| LP(O2) | 1.9306 | — | sp1.56 | s(38.98%)p(60.98%)d(0.04%) | |
| LP(N1) | 1.6359 | — | sp2.21 | s(31.13%)p(68.87%)d(0.01%) | |
| LP(N3) | 1.7106 | — | sp5.15 | s(16.26%)p(83.74%)d(0.00%) | |
Interaction with CT-DNA
The absorption spectra of 2–4 in the absence and presence of CT-DNA (at a constant concentration of complexes) are given in Fig. 7, ESI Fig. S27 and S28.† As seen in the figures, the complexes exhibit intense absorption bands at 260–275 nm assigned to intraligand transitions of the aromatic chromophore. Addition of increasing amounts of CT-DNA results in hypochromism accompanied by a bathochromic shift of about 8 and 7–8 nm for 2 and 3, while hyperchromism is observed for 4 with no shift in the absorption maxima.
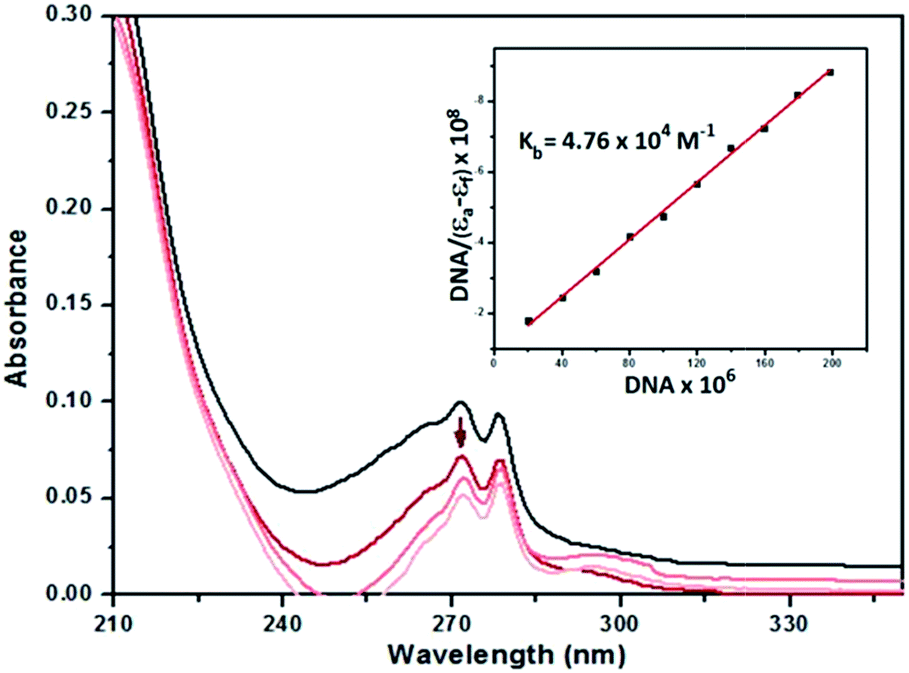 | ||
| Fig. 7 UV spectra of 2 (20 μM) in Tris–HCl buffer in the presence of increasing amounts of CT-DNA (0–200 μM). The arrow indicates decrease in the absorbance on additions of DNA. Inset: linear fit of [DNA]/(εa − εf) vs. [DNA]. | ||
The outcome of the UV titration experiments suggest that all the complexes bind to CT-DNA. The existence of hypochromism for complexes 2 and 3 may be considered as evidence that the complexes bind to DNA via intercalation67 while the opposite effect for 4 suggests surface binding. The magnitude of the binding strength of the complexes with CT-DNA can be estimated through the binding constant. The Kb values (Table 6) suggest a moderate to strong binding of the complexes to CT-DNA with complex 2 exhibiting the highest Kb. The Kb values of the complexes (Table 6) are lower than the classical intercalator EtBr binding affinity for CT-DNA, (Kb = 1.23 (±0.07) × 105 M−1).69
| Complex | UV titration | EtBr exchange | |||
|---|---|---|---|---|---|
| K b (M−1) × 10−4 | K SV (M−1) × 10−4 | K app (M−1) × 10−6 | K F (M−1) × 10−5 | n | |
| 2 | 4.76 ± 0.08 | 3.17 ± 0.02 | 5.42 | 2.30 ± 0.09 | 1.22 |
| 3 | 3.45 ± 0.10 | 2.39 ± 0.04 | 4.21 | 1.06 ± 0.03 | 1.17 |
| 4 | 2.22 ± 0.15 | 1.53 ± 0.06 | 3.04 | 0.21 ± 0.05 | 1.03 |
Competitive studies with ethidium bromide
The complexes show no fluorescence at room temperature in solution or in the presence of CT-DNA, and their binding to DNA cannot be directly predicted through the emission spectra. On the other hand, free EtBr does not exhibit significant emission in buffer solution due to fluorescence quenching by the solvent molecules, while its fluorescence intensity is highly enhanced upon addition of CT-DNA, due to its strong intercalation with DNA base pairs. Thus, competitive EtBr binding studies were carried out so as to investigate the binding of each complex with DNA.The addition of 2–4 to the solutions of CT-DNA pretreated with EtBr caused obvious reduction in emission intensity, indicating that the complexes compete with EtBr in binding to CT-DNA (Fig. 8 and ESI Fig. S29 and S30†). The binding constants (Kapp, KF), quenching constants (KSV), number of binding sites (n) are listed in Table 6. The KSV values for the complexes are in the range of 1.53 × 104 to 3.17 × 104 M−1, respectively, suggesting that EtBr molecules are effectively displaced from the DNA binding sites. The KF values calculated from Scatchard plots (ESI Fig. S31†) are found to be 5.42 × 105, 4.21 × 105, and 3.04 × 105 for 2–4, respectively, indicating that 2 presents the highest binding propensity and the order of binding strength of the complexes to CT-DNA is 2 > 3 > 4, being the same as observed in the UV titration studies. The n values around 1.0 well correspond to a 1:1 molar ratio between the complexes and CT-DNA.
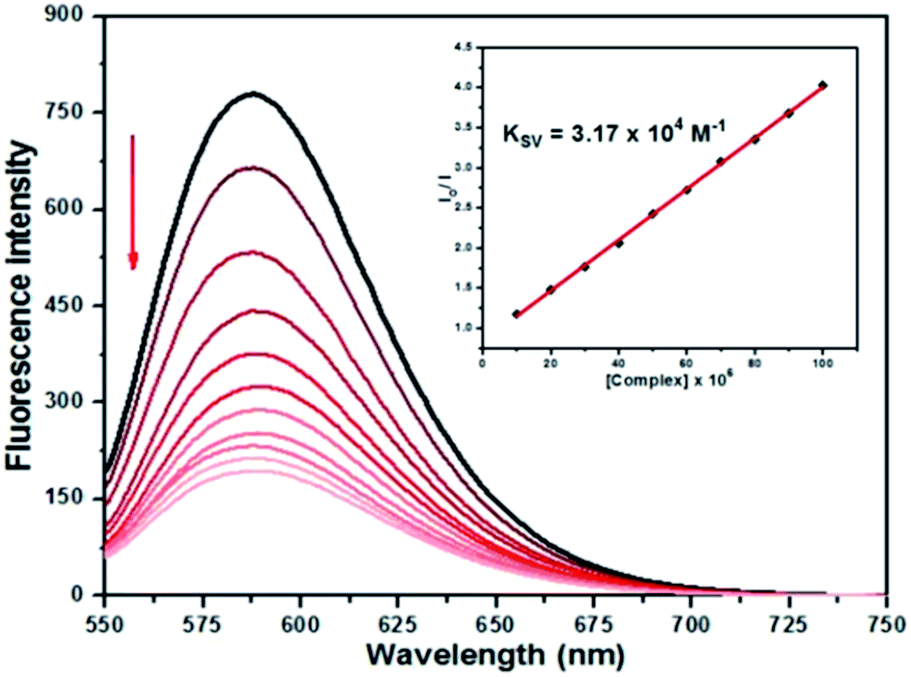 | ||
| Fig. 8 Emission spectra of EtBr-bound DNA solutions in the absence and presence of increasing concentrations of 2 (10–100 μM) in Tris–HCl. [EtBr] = 20.0 μM, [DNA] = 20.0 μM. The arrow shows the change in intensity upon increasing amounts of the complex. Inset: Stern–Volmer plot of the fluorescence data. | ||
Molecular docking
AutoDock has been used for rigid molecular docking studies with DNA duplex of sequence d(CGCGAATTCGCG)2 (1DNE) dodecamer and an octamer d(GAAGCTTC)2 (1DSC). Complexes 2–4 were successively docked into DNA to predict the appropriate binding site and preferred orientation inside the DNA duplex. The lowest-energy conformations of the docked structures of 2–4 are illustrated in Fig. 9 and the magnified views in ESI Fig. S32.† The planarity of the ambim moiety is compatible for π–π stacking interactions with the DNA base pairs and indeed, it is seen that complexes 2 and 3 intercalates in the G-C rich region of the DNA structure. On the other hand, the minimum energy docked pose of 4 reveals that it fits into the curved contour of the targeted DNA in the minor groove. The large GSH moiety in 4 increases its steric bulk, possibly preventing any intercalative interaction. Complex 4 is mainly stabilized by hydrogen bonding through the NH groups of the GSH moiety while the interaction of 2 and 3 with DNA is dominated by π–π interactions with the benzimidazole ring of ambim ligand. On the whole, these interactions (ESI Table S6†) contribute significantly to the stabilization of each complex within the DNA duplex. The binding free energies of the docked structures were computed to be −30.25, −30.12 and −28.45 kJ mol−1 for 2, 3 and 4 respectively. The calculated values are consistent with the trend obtained from the absorption titration and fluorescence quenching studies of CT-DNA (Table 6) and indicate greater binding affinity of 2 relative to the other complexes.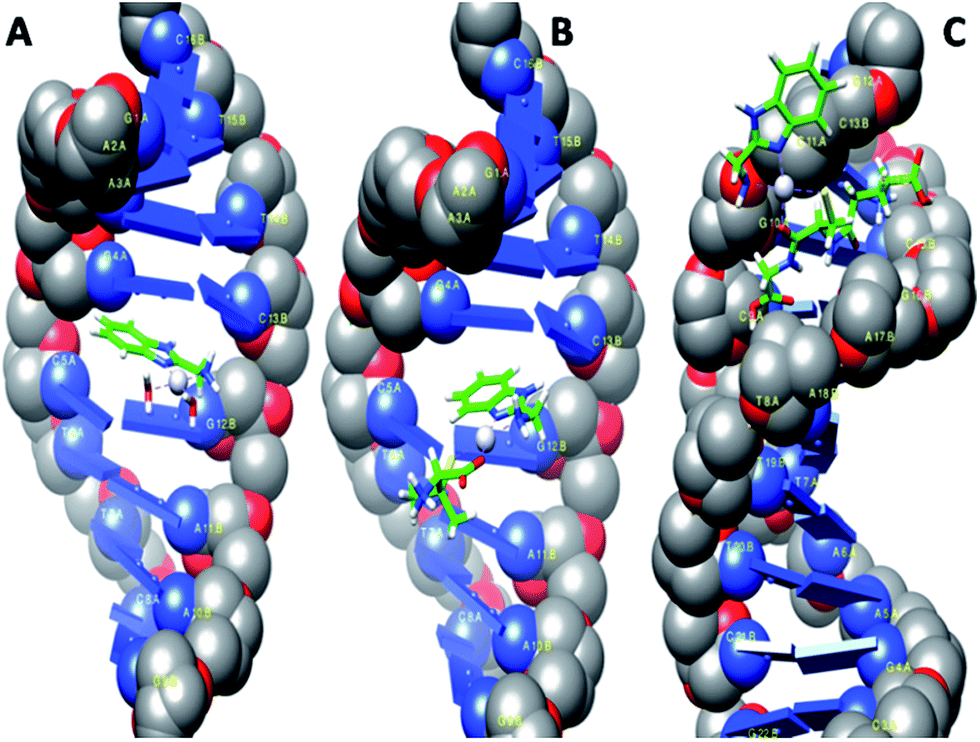 | ||
| Fig. 9 Computational docking models illustrating the interactions of 2 (A) and 3 (B) with 1DNE and of 4 (C) with 1DSC. | ||
DNA cleavage study
The chemical nuclease activity of 2–4 was studied using pCDNA3 plasmid DNA by agarose gel electrophoresis in the absence of any external effect such as UV light, reducing and oxidizing agents. The efficiency of the complexes was assessed by their ability to convert the closed circular supercoiled form of the DNA (form I) to the open circular relaxed or nicked form (form II) or the linear form (form III). The complexes were found to promote the cleavage of the plasmid DNA from supercoiled form to nicked form (lanes 2, 4 and 6, ESI Fig S33†). The mobility of the bands decreases with increasing concentration of the complexes. Moreover, bands for both form I and II are depleted and appear more faint at higher concentration (100 μM) of the complexes (lanes 3, 5 and 7) indicating degradation of plasmid DNA into undetectable single-stranded fragments. The smear that appears in the lanes 2–7 may result from the displacement of EtBr used to stain DNA in the gel by the complexes due to strong interaction. From the gel picture, it can be concluded that complex 2 shows considerably higher DNA cleavage activity than 3 and 4.Cytotoxicity of complexes 2–4 and IC50 values
The encouraging results obtained from the DNA binding studies have directed us to investigate the cell growth inhibition of the synthesised complexes. In vitro cytotoxicity tests were performed using the MTT assay, following exposure of three tumor cell lines: human cervical cancer (HeLa), non-small cell lung carcinoma (A549) and hepatocellular liver carcinoma (HepG2) to the synthesized complexes at increasing concentrations for 48 h. Data for cisplatin and the spectator ligand, ambim, under the same conditions have been also reported for comparison. These complexes exhibit dose-dependent growth inhibitory effect against the tested cell lines and the concentration–response curves are shown in Fig. 10.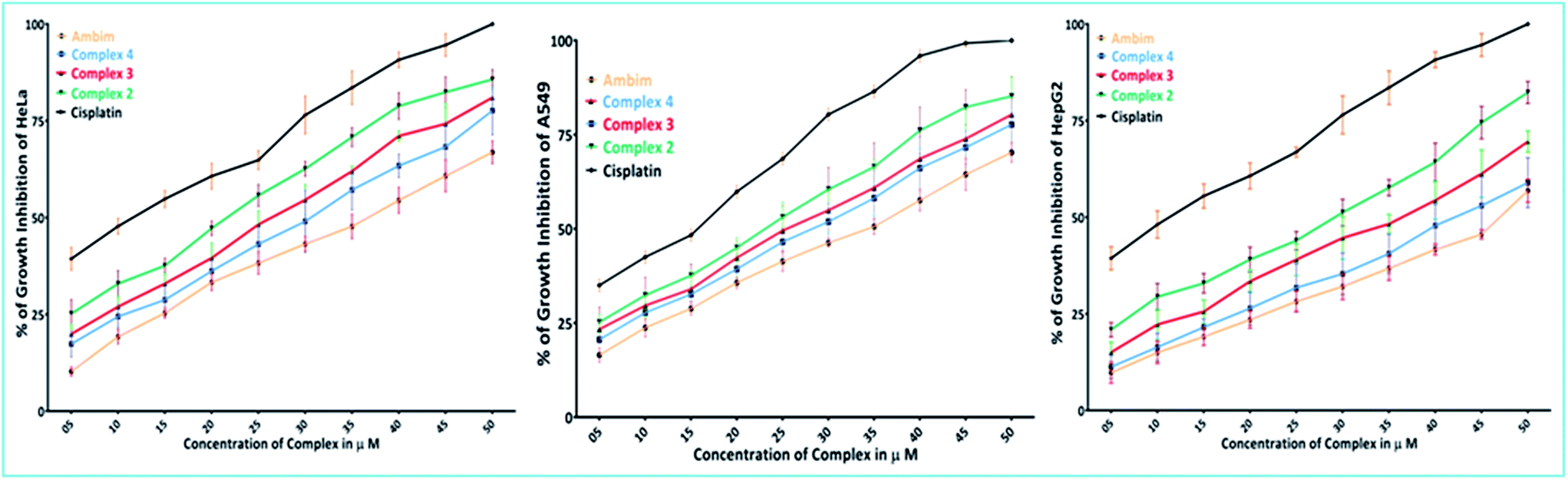 | ||
| Fig. 10 Percentage of growth inhibition of HeLa, A549 and HepG2 cells in presence of ambim, complex 2–4 and cisplatin at concentrations from 5.0 μM to 50.0 μM. All determinations are expressed as percentage of the control (untreated cells). | ||
The IC50 values derived from dose–response curves are summarised in Table 7 and they are comparable to those of cisplatin. Among the compounds studied here, complex 2 is the most cytotoxic in all the three cell lines tested while 4 is the least.
| Compounds | IC50 ± SD, μM | ||
|---|---|---|---|
| HeLa | A549 | HepG2 | |
| Ambim | 37 ± 0.9 | 36 ± 0.8 | 46 ± 1.0 |
| 2 | 22 ± 0.6 | 24 ± 0.7 | 29 ± 0.4 |
| 3 | 26 ± 0.5 | 26 ± 1.1 | 37 ± 0.6 |
| 4 | 31 ± 1.0 | 29 ± 0.4 | 43 ± 0.8 |
| Cisplatin | 12 ± 0.9 | 16 ± 0.6 | 13 ± 1.2 |
The complexes were more effective in arresting the growth of HeLa and A549 than HepG2. It is noteworthy that metal-free ambim was significantly active against all the cell lines at the 48 h time point.
Cell morphology observation
Fig. 11 shows the morphological changes of untreated and treated HepG2 cells observed under a light inverted microscope. The control cells were irregular confluent aggregates with rounded and polygonal cells. The cells treated with complexes 2–4 (B–D), ambim (E) and cisplatin (F) appeared to shrink, became spherical in shape and the cell spreading patterns were restricted (Fig. 11B–F). There is a notable reduction in cell number brought about by the complexes, though the effect is more pronounced for cisplatin. Similar observations were made for HeLa (ESI Fig. S34†) and A549 (ESI Fig. S35†) cells after being treated with the complexes. | ||
| Fig. 11 Microscopic images (A) untreated HepG2 cells, (B) treated with 10 μM of complex 2, (C) complex 3, (D) complex 4, (E) ambim and (F) cisplatin. | ||
Assessment of viability and ROS levels in mice liver and lung cells
It is now well-known that cisplatin exposure results in oxidative stress in normal cells which leads to its non-specific toxicity.7 Excessive reactive oxygen species (ROS) are generated which damage cellular proteins, lipids and DNA, ensuing cell death or apoptosis. Hence, though complexes 2–4 demonstrate marked anti-cancer activity, an obvious question that could be raised is whether they are toxic to normal cells. To have an answer to this, we tested their cytotoxic effect on normal mice liver and lung cells. It is interesting to note that the complexes did not show any adverse effect on normal cells as evidenced from the MTT assay (Fig. 12 and ESI Fig. S36†).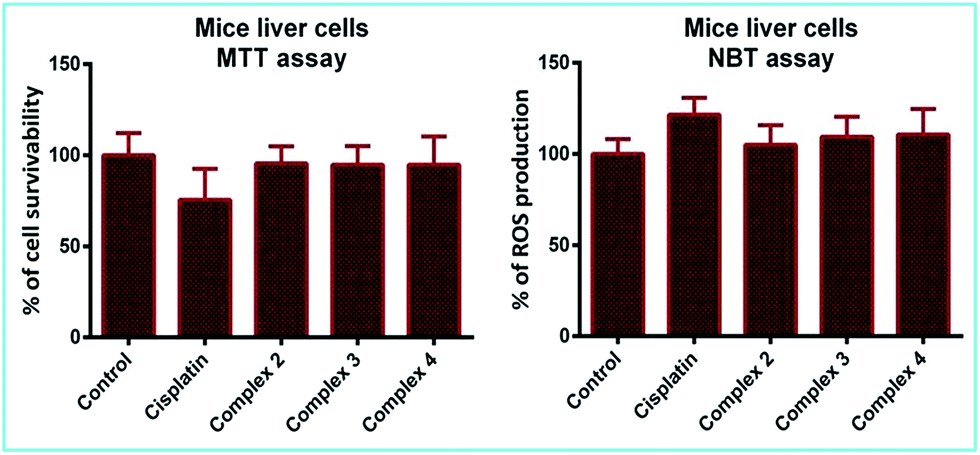 | ||
| Fig. 12 Percentage of cell survivability and ROS production in mice liver cells after 4 h incubation with 50 μM of cisplatin and complexes 2–4. | ||
Besides evaluating cell viability, we also checked the level of superoxides or ROS generated in the afore-mentioned cells, which is an important parameter to determine cellular stress. The results from the NBT assay indicate that ROS production in the cells treated with complexes 2–4 remains almost the same as compared to the control and also lower than those treated with cisplatin (Fig. 12 and ESI Fig. S36†). This suggests that the complexes are not toxic to normal cells, which is a crucial criterion for a practical anti-cancer agent.
Conclusions
The dichloro Pt(II) complex 1, bearing the main ligand, 2-aminomethylbenzimidazole, its diaqua analogue, 2 and the thiol-substituted complexes 3 and 4 have been synthesised and characterized in detail by elemental analysis, NMR, IR and ESI-MS. DL-Penicillamine and glutathione coordinates to Pt(II) centre in complex 2 through S and O donors to form six membered complexes 3 and 4via two consecutive steps. The low values of the enthalpies of activation (ΔH‡1 and ΔH‡2) and large negative values of entropies of activation (ΔS‡1 & ΔS‡2) for both reactions imply a good degree of ligand participation in associative path of the T. S. The trend in the reactivity of the ligands is also reflected in the activation enthalpies of the reactions. The higher nucleophilicity of GSH leads to greater stabilization of the T.S. and hence requires a smaller activation enthalpy as compared to DL-pen. DFT calculations determine the global minimum optimized structures of complexes 2–4, which reveals few important bond distances (Å) and bond angles (°) close to reported values. Electronic spectra calculated by TD-DFT is in good agreement with the observed UV-vis spectra.Based on the data from absorption and fluorimetric competitive titrations, it can be concluded that the complexes bind strongly to CT-DNA with different binding affinities. The calculated binding constants of the complexes with DNA showed that 2 has the highest binding affinity to DNA. Intercalative binding for complexes 2 and 3, and minor groove binding for 4 was further validated by molecular docking studies. These water soluble complexes are also efficient DNA cleavers and exhibit considerable cytotoxicity on HeLa, A549 and HepG2 cell lines. Based on the IC50 values, the potency of the complexes can be ordered as 2 > 3 > 4. On all the three cell lines, complex 2 shows 82–85% growth inhibition at higher concentration (50 μM) as compared to cisplatin. Most importantly, the complexes were found to generate lower levels of ROS and better viability for normal cells than cisplatin.
Further studies can be undertaken to assess their pharmacological properties in vivo and elucidate the definite mechanism of their bioactivity. The results presented here may be beneficial in understanding the interactions of Pt(II) complexes with intracellular thiols and DNA and thereby useful in the development of superior anticancer agents.
Acknowledgements
The authors are thankful to National Institute of Technology Durgapur, W.B. 713209, India and DST, Govt. of India (Project no. SB/EMEQ-028/2013) for providing the necessary assistance and financial support to carry out this work. Ishani Mitra and Subhajit Mukherjee are also thankful to DST Inspire, Govt. of India for research fellowship. Sandip Mukherjee is thankful to DST, SERB N-PDF for research fellowship. We are grateful to the reviewers for their valuable suggestions.References
- B. Rosenberg, in Cisplatin: Chemistry and Biochemistry of a Leading Anticancer Drug, Verlag Helv. Chim. Acta, ed. B. Lippert, Wiley-VCH. Zurich, Weinheim, Germany, 1999, p. 3 Search PubMed.
- M. J. Hannon, Pure Appl. Chem., 2007, 79(12), 2243 CrossRef CAS.
- S. Dasari and P. Bernard Tchounwou, Eur. J. Pharmacol., 2014, 740, 364 CrossRef CAS PubMed.
- L. Kelland, Nat. Rev. Cancer, 2007, 7, 573 CrossRef CAS PubMed.
- Z. H. Siddik, Oncogene, 2003, 22, 7265 CrossRef CAS PubMed.
- C. A. Rabik and M. E. Dolan, Cancer Treat. Rev., 2007, 33, 9 CrossRef CAS PubMed.
- R. Marullo, E. Werner, N. Degtyareva, B. Moore, G. Altavilla, S. S. Ramalingam and P. W. Doetsch, PLoS One, 2013, 8(11), 81162 Search PubMed.
- Q. Yang, H. Xiao, J. Cai, Z. Xie, Z. Wang and X. Jing, RSC Adv., 2014, 4, 61124 RSC.
- Q. Yang, R. Qi, J. Cai, X. Kang, S. Sun, H. Xiao, X. Jing, W. Li and Z. Wang, RSC Adv., 2015, 5, 83343 RSC.
- H. Xiao, L. Yan, Y. Zhang, R. Qi, W. Li, R. Wang, S. Liu, Y. Huang, Y. Li and X. Jing, Chem. Commun., 2012, 48, 10730 RSC.
- H. Xiao, J. F. Stefanick, X. Jia, X. Jing, T. Kiziltepe, Y. Zhang and B. Bilgicer, Chem. Commun., 2013, 49, 4809 RSC.
- R. Qi, H. Xiao, S. Wu, Y. Li, Y. Zhang and X. Jing, J. Mater. Chem. B, 2015, 3, 176 RSC.
- H. Song, W. Li, R. Qi, L. Yan, X. Jing, M. Zheng and H. Xiao, Chem. Commun., 2015, 51, 11493 RSC.
- R. Wang, H. Xiao, H. Song, Y. Zhang, X. Hu, Z. Xie, Y. Huang, X. Jing and Y. Li, J. Mater. Chem., 2012, 22, 25453 RSC.
- R. Wang, X. Hu, H. Xiao, Z. Xie, Y. Huang and X. Jing, J. Mater. Chem. B, 2013, 1, 744 RSC.
- J. K. Barton and S. J. Lippard, Biochemistry, 1979, 18, 2661 CrossRef CAS PubMed.
- B. M. Zeglis, V. C. Pierre and J. K. Barton, Chem. Commun., 2007, 4565 RSC.
- D. M. Fisher, R. R. Fenton and J. R. Aldrich-Wright, Chem. Commun., 2008, 5613 RSC.
- A. C. G. Hotze, E. P. L. van der Geer, H. Kooijman, A. L. Spek, J. G. Haasnoot and J. Reedijk, Eur. J. Inorg. Chem., 2005, 2648 CrossRef CAS.
- S. Komeda, T. Moulaei, K. K. Woods, M. Chikuma, N. P. Farrell and L. D. Williams, J. Am. Chem. Soc., 2006, 128, 16092 CrossRef CAS PubMed.
- D. L. Ma, T. Y. T. Shum, F. Zhang, C. M. Che and M. Yang, Chem. Commun., 2005, 4675 RSC.
- A. A. Spasov, I. N. Yozhitsa, L. I. Bugaeva and V. A. Anisimova, Pharm. Chem. J., 1999, 33, 232 CrossRef CAS.
- G. L. Gravatt, B. C. Baguley, W. R. Wilson and W. A. Denny, J. Med. Chem., 1994, 37, 4338 CrossRef CAS PubMed.
- J. P. Lalezari, J. A. Aberg, L. H. Wang, M. B. Wire, R. Miner, W. Snowden, C. L. Talarico, S. Shaw, M. A. Jacobson and W. L. Drew, Antimicrob. Agents Chemother., 2002, 46, 2969 CrossRef CAS PubMed.
- D. Labrecke, S. Lamothe, M. Courchesne, L. Chan, G. Attardo and K. Meerovitch, Preparation of thiophene-2,5-dicarboxamides and furan-2,5-dicarboxamides useful in the treatment of cancer, US Pat., 91063, 1998.
- W. H. Miller, W. E. Bondinell, T. W. Ku, R. M. Keenan, J. M. Samanen, C. Kwon, F. E. Ali and M. A. Lago, Benzimidazole derivatives and analogs as vitronectin receptor antagonists, US Pat., 9366, 1995.
- M. A. Jakupec, M. Galanski and B. K. Keppler, Rev. Physiol., Biochem. Pharmacol., 2003, 146, 1 CAS.
- J. Reedijk, Chem. Rev., 1999, 99, 2499 CrossRef CAS PubMed.
- K. S. Lovejoy and S. J. Lippard, Dalton Trans., 2009, 48, 1065 Search PubMed.
- S. H. van Rijt and P. J. Sadler, Drug Discovery Today, 2009, 14, 1089 CrossRef CAS PubMed.
- R. W. Hay and A. K. Basak, J. Chem. Soc., Dalton Trans002E, 1982, 1819 RSC.
- A. A. El-Sherif, in Coordination Chemistry of Palladium(II) Ternary Complexes with Relevant Biomolecules, Stoichiometry and Research – The Importance of Quantity in Biomedicine, ed. A. Innocenti, InTech, 2012, ch. 4, pp. 79–120 Search PubMed.
- J. A. Weyh and R. E. Hamm, Inorg. Chem., 1969, 8, 2298 CrossRef CAS.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2009 Search PubMed.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 299 CrossRef CAS.
- P. J. Hay and W. R. Wadt, J. Chem. Phys., 1985, 82, 284 CrossRef.
- R. Ditchfield, W. J. Hehre and J. A. Pople, J. Chem. Phys., 1971, 54, 724 CrossRef CAS.
- W. Hehre, J. R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257 CrossRef CAS.
- D. E. Glendening, A. E. Reed, J. E. Carpenter and F. Weinhold, NBO, Version 3.1 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 3, 455 Search PubMed.
- C. G. Ricci and P. A. Netz, J. Chem. Inf. Model., 2009, 49, 1925 CrossRef CAS PubMed.
- A. M. Pyle, J. P. Rehmann, R. Meshoyrer, C. V. Kumar, N. J. Turro and J. K. Barton, J. Am. Chem. Soc., 1989, 111, 3051 CrossRef CAS.
- O. Stern and M. Volmer, Phys. Z., 1919, 20, 183 CAS.
- M. Lee, A. L. Rhodes, M. D. Wyatt, S. Forrow and J. A. Hartley, Biochemistry, 1993, 32, 4237 CrossRef CAS PubMed.
- J. Min, X. M. Meng-Xia, Z. Dong, L. Yuan, L. Xiao-Yu and C. Xing, J. Mol. Struct., 2004, 692, 71 CrossRef CAS.
- R. I. Freshney, Culture of animal cells: A manual of basic technique, Wiley-Liss, NY, 5th edn, 1987, p. 200 Search PubMed.
- R. W. Masters, Animal cell culture: Cytotoxicity and viability assays, 3rd edn, 2000, vol. 23, p. 202 Search PubMed.
- D. Garmann, A. Warnecke, G. V. Kalayda, F. Kratz and U. Jaehde, J. Controlled Release, 2008, 131, 100 CrossRef CAS PubMed.
- Y. C. Sasaki and A. Passaniti, BioTechniques, 1998, 24, 1038 Search PubMed.
- F. Gao, H. Chao, F. Zhou, Y. X. Yuan, B. Peng and L. N. Ji, J. Inorg. Biochem., 2006, 100, 1487 CrossRef CAS PubMed.
- A. A. Holder, S. Swavey and K. J. Brewer, Inorg. Chem., 2004, 43, 303 CrossRef CAS PubMed.
- G. Cervantes, V. Moreno, E. Molins and M. Quiros, Polyhedron, 1998, 17, 3343 CrossRef CAS.
- C. T. Yang, B. Moubaraki, K. S. J. Murray and J. Vittal, Dalton Trans., 2003, 880 RSC.
- B. K. Singh, P. Mishra and B. S. Garg, Spectrochim. Acta, Part A, 2007, 67, 719 CrossRef PubMed.
- G. B. Deacon, F. Huber and R. J. Phillips, Inorg. Chim. Acta, 1985, 104, 41 CrossRef CAS.
- R. M. Silverstein, O. G. Bassler and T. C. Morrill, Spectrophotometic Identification of Organic Compounds, Wiley, New York, 1974 Search PubMed.
- C. J. Pouchert and J. Behnke, The Aldrich Library of 13C and 1H FTNMR Spectra, Aldrich Chemical Company, Wisconsin, 1993, vol. I, p. 273 Search PubMed.
- N. S. Sisombath, F. Jalilehvand, A. C. Schell and Q. Wu, Inorg. Chem., 2014, 53, 12459 CrossRef CAS PubMed.
- J. Arpalahti and B. Lippert, Inorg. Chem., 1990, 29, 104 CrossRef CAS.
- W. Shen, R. D. Schnebeck, E. Freisinger and B. Lippert, Dalton Trans., 2008, 4044 RSC.
- K. Sakai, Y. Konno, N. Takayama and S. Takahashi, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2004, 60, 255 Search PubMed.
- E. Colacio, R. Cuesta, M. Ghazi, M. A. Huertas, J. M. Moreno and A. Navarrete, Inorg. Chem., 1997, 36, 1652 CrossRef CAS PubMed.
- R. Wysokinśki, K. Hernik, R. Szostak and D. Michalska, Chem. Phys., 2007, 333, 37 CrossRef.
- F. Dimiza, F. Perdih, V. Tangoulis, I. Turel, D. P. Kessissoglou and G. Psomas, J. Inorg. Biochem., 2011, 105, 476 CrossRef CAS PubMed.
- A. Tarushi, C. P. Raptopoulou, V. Psycharis, A. Terzis, G. Psomas and D. P. Kessissoglou, Bioorg. Med. Chem., 2010, 18, 2678 CrossRef CAS PubMed.
- H. Wu, Y. Zhang, C. Chen, J. Zhang, Y. Bai, F. Shi and X. Wang, New J. Chem., 2014, 38, 3688 RSC.
- A. Dimitrakopoulou, C. Dendrinou-Samara, A. A. Pantazaki, M. Alexiou, E. Nordlander and D. P. Kessissoglou, J. Inorg. Biochem., 2008, 102, 618 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17788c |
| This journal is © The Royal Society of Chemistry 2016 |