
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of fluids in high-pressure polymorphism of drugs: different behaviour of β-chlorpropamide in different inert gas and liquid media
B. A.
Zakharov
*ab,
Y. V.
Seryotkin
abc,
N. A.
Tumanov
de,
D.
Paliwoda
f,
M.
Hanfland
f,
A. V.
Kurnosov
g and
E. V.
Boldyreva
*a
aInstitute of Solid State Chemistry and Mechanochemistry SB RAS, Kutateladze Str. 18, Novosibirsk 630128, Russia. E-mail: b.zakharov@yahoo.com; eboldyreva@yahoo.com
bNovosibirsk State University, Pirogova Str. 2, Novosibirsk 630090, Russia
cV.S. Sobolev Institute of Geology and Mineralogy SB RAS, Ac. Koptyuga ave. 3, Novosibirsk 630090, Russia
dInstitute of Condensed Matter and Nanosciences, Université catholique de Louvain, Place L. Pasteur 1, Louvain-la-Neuve 1348, Belgium
eUniversité de Namur, Rue de Bruxelles 61, Namur 5000, Belgium
fEuropean Synchrotron Radiation Facility, Avenue des Martyrs 71, Grenoble 38000, France
gBayerisches Geoinstitut, Universität Bayreuth, Universitätsstraße 30, Bayreuth D-95447, Germany
First published on 21st September 2016
Abstract
The hydrostatic compression of β-chlorpropamide gives different high-pressure phases, depending on the choice of pressure-transmitting fluid (paraffin, neon and helium). This is particularly surprising as none of these fluids interact obviously with the solid at ambient pressure. This phenomenon is not related to dissolution and recrystallization, in contrast to what has been previously observed for β-chlorpropamide in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 pentane–isopentane mixture.
:1 pentane–isopentane mixture.
Introduction
The last decade has seen an explosive increase in the number of publications on the effect of hydrostatic pressure on organic crystals.1–8 One of the reasons for this is that new polymorphs can be formed. This is of fundamental interest for the design of crystal structures and understanding the mechanisms of crystallisation and solid-state transformations. High-pressure polymorphism is also of practical importance, with new phases being used as obtained or as seeds for subsequent mass-crystallization. These applications are particularly relevant if the high-pressure phase can be preserved on decompression. Polymorph discovery is of special interest to the pharmaceutical industry,7,9–11 since different polymorphs differ not only in their physical and biological properties, but are also separate legal entities, are patentable and are thus important for intellectual property.12,13It is generally not possible to predict which phase will be formed at a selected (T, P) point based solely on a thermodynamic phase diagram. Instead, there exists a complex interplay of nucleation and growth kinetics, alongside thermodynamics, that leads to unpredictable results.14 As a result of the kinetic control of nucleation and nuclei growth,14 different phases can form depending on the choice of the starting polymorph,15–19 the compression/decompression protocol,20–23 and the choice of hydrostatic medium.24–28 The latter is of particular interest to the present study.
Hydrostatic conditions cannot be achieved without a hydrostatic medium – a fluid phase, i.e. a gas or a liquid. In high-pressure mineralogy, the importance of selecting a pressure-transmitting fluid is well understood. This has become particularly important in relation to microporous materials, zeolites, which are characterized by an “open” system of interconnected channels and cavities.29 For example, when present in the hydrostatic medium (even in trace quantities), molecules of H2O can enter zeolite channels upon compression; this process is reversible.30 The so-called “pressure-induced hydration”31 can cause either volume increase (abrupt as in ref. 31 and 32 or gradual as in ref. 33), or simply a decrease in compressibility as compared with the non-hydrated species, due to the site occupancy increase of the already existing water sites.34,35 It is important to note, however, that this pressure-induced hydration effect is not always observed.36,37 Gasses used as pressure-media are generally inert with respect to minerals, but gases such as hydrogen or helium can also penetrate into the solid. This can result in a decreased compressibility of the material,38–41 or also in the structural transformations, as for cristobalite,42 or vitreous silica43 compressed in helium. Despite these effects being known for minerals, the choice of pressure-transmitting medium (PTM) for high-pressure research of organic molecular crystals has been rather arbitrary. Most commonly, a PTM is chosen for practical reasons, often simply based on which are physically available in the laboratory. Gas loading is not widely available, and similarly, not all groups have the facilities to work with low boiling point fluids, such as pentane–isopentane.44 In practice, a fluid is selected not only for availability, but (i) to ensure hydrostatic conditions are maintained up to the desired pressure point, and (ii) to avoid obvious chemical interaction with the immersed solid, while simultaneously avoiding its dissolution. For the former, there exist large repertoires of data summarizing the hydrostaticity limits of various fluids.45,46 Recent studies with precise pressure measurements across multiple sites in a pressure cell show that a notable loss of ideal hydrostaticity (thus possible shear stresses in the medium) occur at considerably lower pressures than was previously expected from either the broadening of X-ray diffraction peaks or from the measurement of a single pressure point in the cell.45 In interactions of the PTM with the sample, it is very difficult (if not impossible) to predict a priori whether the solubility of a substance will change with pressure. This is particularly problematic when a compound is insoluble in the PTM under ambient conditions, but dissolves under pressure. It is also difficult to predict surface interactions between a solvent and the crystal material, an interaction that may increase with pressure. One must therefore always consider the possibilities of high-pressure dissolution, re-crystallisation, and solvate formation at elevated pressures.
A number of studies dedicated specifically to the role of fluids in pressure-induced solid-state transformations have been published.6,21,23–28,47–83 Fluids have been documented to reversibly penetrate into crystal structures to form inclusion compounds, especially if the solid structure has channels or large cavities, such as MOFs47–55 or zeolites.30–35 Additionally, solids have been reported to recrystallise into solvates at high pressure.6,21,23,28,56–70 It was also shown that some solid-state pressure-induced phase transitions can be solvent-assisted, i.e. observed in some fluids, and non-observed in others.24–27 This phenomenon is also known for ambient-pressure crystallisation.71–82 Transformations in liquids, in which solids can dissolve only under pressure – often recrystallising as a new high-pressure phase – are sometimes compared with those in inert fluids (i.e. those in which samples do not dissolve under pressure, so that real solid state transformations are possible). A systematic investigation into the variety of possible effects fluids may have on pressure-induced transformations of solids is just emerging. To the best of our knowledge, there are currently no documented examples that compare the effect of pressure on the same solid phase when immersed in different non-dissolving fluids.
The aim of the present work was to compare the effect of pressure on a selected organic crystal using several “inert” hydrostatic fluids, none of which can visibly dissolve the solid under ambient conditions. The present work investigates chlorpropamide, (4-chloro-N-(propylamino-carbonyl)benzenesulfonamide, C10H13ClN2O3S) (Fig. 1), an antidiabetic drug which is prone to forming new polymorphs84,85 under ambient and high pressures.25,26,86–89 The α-polymorph, which is the stable form under ambient conditions,90 undergoes at least one phase transition on increasing pressure to give a high-pressure phase, α′-polymorph.26 This high pressure phase is the same as that obtained when the PTM used is either a saturated ethanol solution (recrystallisation possible)26 or a 1:1 pentane–isopentane mixture (no visible dissolution).89 The same phase transition seems to also take place in dry powder samples without the addition of any pressure-transmitting fluids,25 albeit at a considerably slower rate. This has led to the suggestion that the high-pressure transformation in chlorpropamide is kinetically hindered and thus solvent-assisted.25 When different polymorphs (α-, β-, γ-, δ-forms) are compressed in pentane–isopentane, different high-pressure phases are formed at different pressures.89 At the same time, when compressing a sample of the β-polymorph in pentane–isopentane (1:1), different high-pressure phases are formed, depending on the presence of other chlorpropamide polymorphs as seeds.89 This suggests that the pressure-induced structural transformations of the β-polymorph are related to recrystallisation from pentane–isopentane at high pressure, even though this solvent does not visibly dissolve any of the polymorphs at ambient pressure, or the α-, γ-, and δ-forms at high pressure.89 This system therefore offered an intriguing opportunity to compare the effect of an inert liquid (paraffin) with that of inert gases (Ne, He) on the structural transformations in the β-polymorph (Fig. 2). These results could then be compared with those observed on compression in a 1:1 pentane–isopentane mixture, in which the sample starts dissolving as pressure increases.89
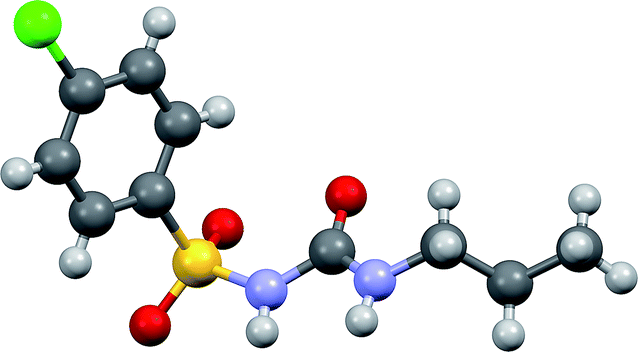 | ||
| Fig. 1 Molecular structure of chlorpropamide. Carbon atoms – grey, hydrogens – light grey, oxygens – red, sulfur – yellow, chlorine – green. | ||
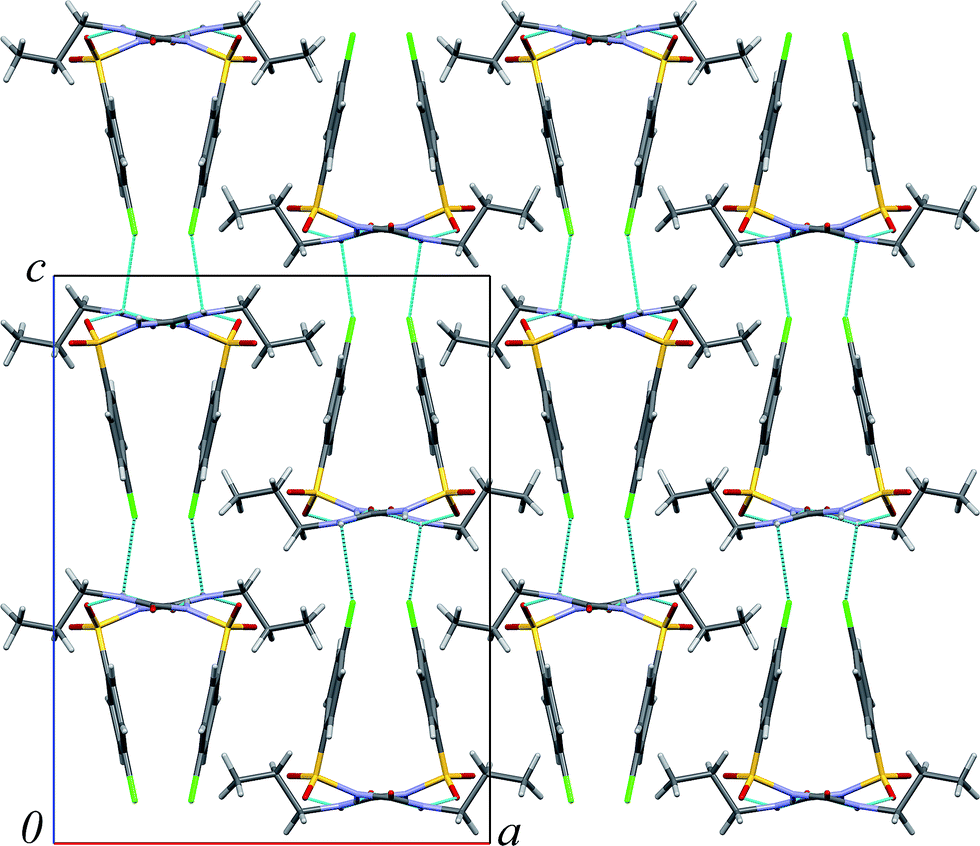 | ||
| Fig. 2 Molecular packing in β-chlorpropamide. Hydrogen bonds are shown by blue lines. | ||
Results and discussion
The main results of the experiments are summarised in Table 1. Microphotographs are given for crystals of β-chlorpropamide loaded in a DAC in neon, immediately after compression to the first pressure point (0.6 GPa) and after the phase transformations in neon, Fig. 3.| Medium | A high-pressure phase | SG and cell parameters | Pressure range when formed, GPa | Polycrystalline or single crystal? |
|---|---|---|---|---|
| a Data for compression in pentane–isopentane are from ref. 89 and given for comparison. | ||||
| Ne | α-Polymorph | P212121, 4.9582(4) Å, 26.502(13) Å, 8.9098(7) Å | Between ambient pressure and 0.6 GPa | Several domains in what was originally a perfect single crystal |
| α′-Polymorph | Cell could not be found (reflections are strongly distorted) | Between 2.3 and 2.6 GPa | Strongly distorted crystal split into several large fragments | |
| He | A new phase, βIHP | Monoclinic, 14.349(5) Å, 9.2192(17) Å, 18.84(4) Å, 90.35(8) ° | Between 0.3 and 0.5 GPa | Single crystal |
| A new phase, βIIHP | Monoclinic, 14.041(6) Å, 9.1411(19) Å, 18.71(4) Å, 94.63(11)° | Between 0.7 and 1.0 GPa | Several domains in what was originally a perfect single crystal | |
| Liquid paraffin | A new phase; βIHP | Monoclinic, 14.399(3) Å, 9.2288(18) Å, 18.87(4) Å, 91.61(3)° | Between ambient pressure and 0.1 GPa | Several domains in what was originally a perfect single crystal |
| A new phase; βIIIHP | Triclinic, 14.200(2) Å, 9.2010(17) Å, 18.858(16) Å, 89.45(3)°, 86.66(3)°, 89.980° | Between 0.1 and 0.3 GPa | Several domains | |
| Not identified | Cell could not be found (reflections are strongly distorted) | Between 1.6 and 2.2 GPa | Strongly distorted crystalline domains | |
| 1:1 pentane–isopentane mixture89 |
γ-Polymorph | Monoclinic, 6.1040(5) Å, 8.9243 Å, 12.0304(14) Å, 99.516(8)° | At 0.1–0.2 GPa | Multiple new crystals |
| 1:1 pentane–isopentane mixture, δ- and α-phases also present |
γ + δ | Not measured, crystal growth was followed visually | 0.3–0.5 GPa | γ-Polymorph as new crystals + δ-polymorph seed growing; no growth of the α-polymorph |
| Non-identified phases after transformation of both forms, characterized by Raman spectra | 2.4–3.3 GPa | Single crystals visually preserved | ||
 | ||
| Fig. 3 β-CPA loaded in neon immediately after loading at BGI (0.6 GPa) and after high-pressure experiment at ESRF (6.0 GPa); the sample at the left photo looked the same two days later when examined at ESRF after transport. | ||
Compression in Ne
A single crystal of the β-polymorph was loaded into a DAC in Ne, with a pressure of 0.6 GPa immediately after closing the cell. The loaded DAC was then transferred to ESRF to perform diffraction experiments. The time interval between DAC loading and the diffraction experiments was therefore two days. The initial diffraction data obtained at the synchrotron source corresponded to the α-polymorph; the high-quality single crystal originally loaded had transformed into several singe-crystalline domains. The pressure measured after the transfer of the DAC to ESRF and the diffraction experiment was 0.5 GPa. Unfortunately, it was not technically possible to repeat the loading of a DAC with a crystal of the β-polymorph in Ne and to track any phase changes at pressures below 0.5 GPa. Therefore, it was not possible to determine the phase transition point more precisely, or investigate any kinetics effects, i.e. if the high-pressure transformation takes place after some delay when kept at a certain pressure. On further compression in steps of 0.5 GPa, the α-polymorph (which formed from the original β-form) transformed into another phase. The transition pressure range (2.3–2.6 GPa) was similar to that at which the transformation of the α-form into the high-pressure α′-polymorph was previously observed on compression in saturated ethanol solution26 (2.6–2.9 GPa), or in pentane–isopentane89 (2.4–3.3 GPa). Obviously, the diffraction pattern no longer corresponded to that of α-polymorph. However, the quality of the diffraction data was not sufficient to unambiguously decide whether the new phase was the same α′-polymorph described in ref. 26 based on single-crystal diffraction, or any other phases which are claimed to exist. Unfortunately, many of these phases have not been characterized, even by cell parameters,87 and have formed apparently without PTM. No obvious transitions were observed on further compression up to 6.0 GPa (the last pressure point achieved in the experiment). In an independent experiment, a crystal of the original α-polymorph in Ne, which was compressed immediately to 4.6 GPa, also transformed into a high-pressure phase. As before, it was not possible to unambiguously identify this phase as the α′-polymorph, or another phase, as the crystal was strongly distorted.Compression in He
When compressing a crystal of the β-polymorph in He, pressure was increased in small steps. The diffraction data at pressures below 0.3 GPa corresponded to the orthorhombic β-polymorph. The first changes in cell parameters, which might be a manifestation of a phase transition into a monoclinic phase (designated βIHP), were observed between 0.3 and 0.5 GPa (Fig. 4). Although the crystal was not visibly destroyed, and the β-angle was still very close to 90° (90.35(8)° at 0.5 GPa), the diffraction data could no longer be described using the structure of β-chlorpropamide as a starting model. Attempts to solve the crystal structure or to refine in any of the other previously known models also failed. The cell parameters of the new monoclinic phase did not correspond to the α-polymorph, or to any of the other previously reported chlorpropamide polymorphs. This suggests that the structural transformation must have been significant and a new βIHP-phase has been formed. The next jump-wise change in the value of the β-angle was observed between 0.7 and 1.0 GPa (Fig. 4). The second new high-pressure phase was designated βIIHP; the cell parameters of this phase were very close to those of the βII-chlorpropamide low-temperature phase, which forms on cooling of the β-polymorph to about 257 K.94 Again, although the crystal was not visibly destroyed, attempts to solve the crystal structure of this new phase, or to refine it using the structure of βII-chlorpropamide as a starting model failed, this indicating that structural distortion/rearrangement was very significant.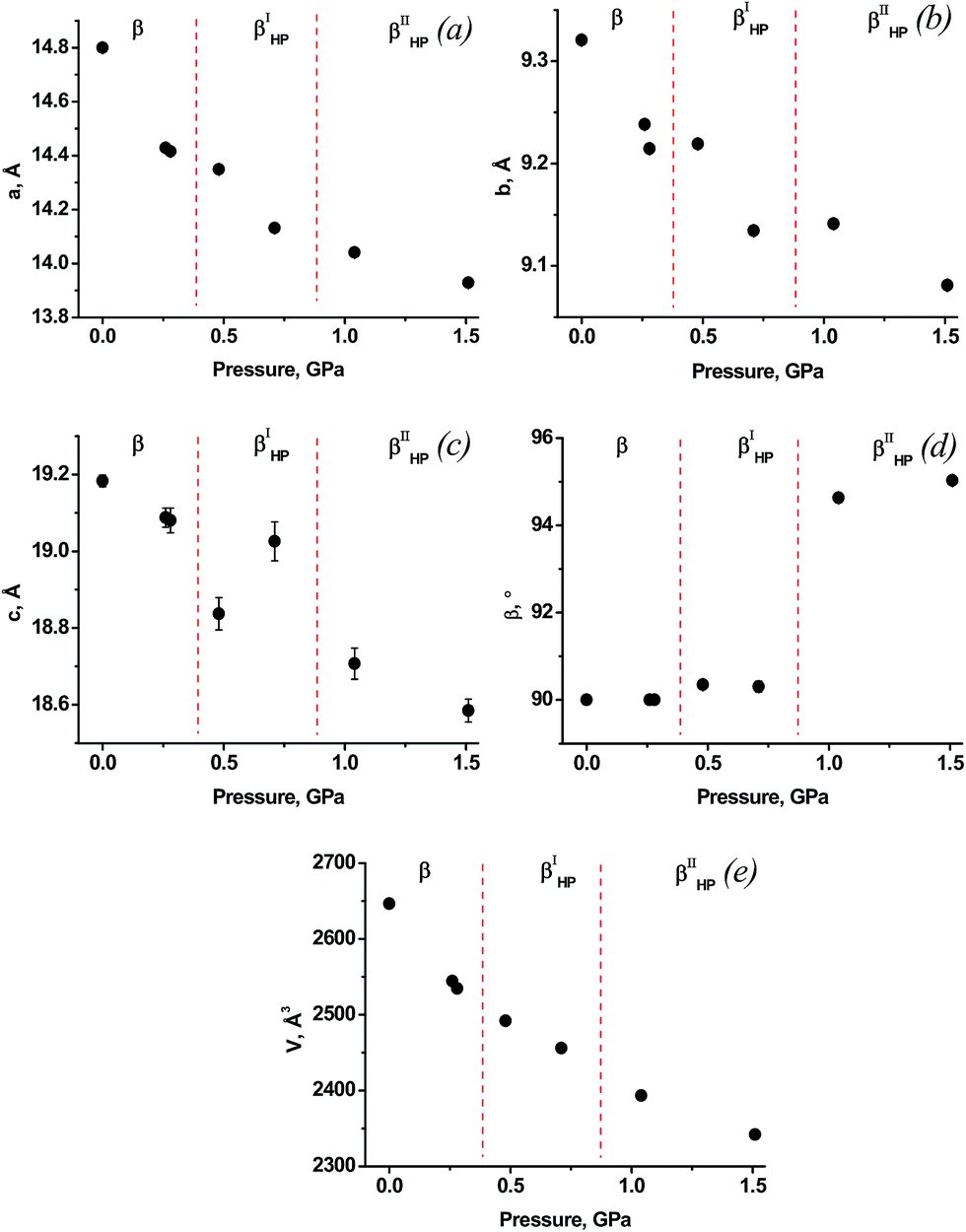 | ||
| Fig. 4 Dependencies of cell parameters on pressure for β-chlorpropamide loaded in He. | ||
So, the response of the crystals of the β-polymorph to pressure was different, depending on the choice of the inert gas. One can suppose that this difference can be a consequence of the difference in the atomic radii of Ne and He. Helium was shown to penetrate into voids in various solids, such as, e.g. crystalline SiO2 (cristobalite),42 arsenolite,92,95 or ice.96 Cristobalite gave a new phase at about 8 GPa which was supposed to have a molar volume of about 30% larger, than cristobalite, suggesting the dissolution of helium atoms in its interstitial voids. As4O6·2He clathrate was shown to appear at the surface layer of arsenolite and penetrate in depth on increasing pressure. Experimental and computational results provide evidence for ordered helium trapping above 3 GPa between adamantane-type As4O6 cages.95
Analysis of voids sizes in β-chlorpropamide (Fig. 2) shows that they are larger than the voids in α-cristobalite93 and arsenolite92 (Table 2). It therefore follows that small He atoms can penetrate into the crystal structure of the β-chlorpropamide, filling the cavities between chlorpropamide molecules (Fig. 2). The changes in cell parameters and volume of the β-chlorpropamide crystals on compression in helium can be caused by two phenomena. First, these changes may be caused by helium inclusions that occur as a result of continuous structural strain (itself being introduced by the hydrostatic compression), or secondly, they may occur as a result of a combination of strain from external hydrostatic compression and internal strain from helium guest atoms. In this respect it may be relevant to note that helium penetration into the interstitial free volume of the glass network of vitreous silica has been documented to induce deformation that accounts for the small apparent compressibility of silica.39
| Pressure transmitting media (further PTM) | He | Ne | Paraffin | Pentane–isopentane |
| Atomic radius/molecule size, Å | 0.31 (1.40 vdW) | 0.38 (1.54 vdW) | >15 | ∼5 |
| Possible dissolution of PTM in β-CPA interstitial voids? | + | ± | − | − |
| Interaction of β-CPA alkyl tales with PTM | − | − | + | + |
| Volume of voids in unit cell of β-CPA available for sphere of a certain atomic radius, Å3 (% of unit cell) | 764.26 (28.9%) | 667.15 (25.2%) | − | − |
| Volume of voids in unit cell of α-cristobalite available for sphere of a certain atomic radius, Å3 | 29.11 (17.0%) | 25.06 (14.6%) | − | − |
| Volume of voids in unit cell of arsenolite available for sphere of a certain atomic radius, Å3 | 312.69 (23.0%) | 297.08 (21.9%) | − | − |
Compression in paraffin
As the third inert fluid, liquid paraffin was used. The values of volume vs. pressure measured are plotted in Fig. 5. Transformation of the β-polymorph into a monoclinic phase with cell parameters similar to those of βIHP-chlorpropamide was observed to have occurred by 0.1 GPa. As pressure was increased to 0.3 GPa, another high-pressure phase was formed, with a diffraction pattern that could be indexed by assuming a triclinic cell that does not correspond to any previously reported crystal structures of chlorpropamide (βIIIHP). Further compression shows that the V(P) curve does not have discontinuities up to 1.6 GPa. On further pressure increase to 2.2 GPa the crystal was destroyed, yielding a polycrystalline phase. Attempts to index the diffraction pattern and find the cell parameters failed, as the reflections were strongly distorted. This is presumably due to structural distortion. This again shows that the pressure-induced transformation must be related to a rather significant structural reorganisation.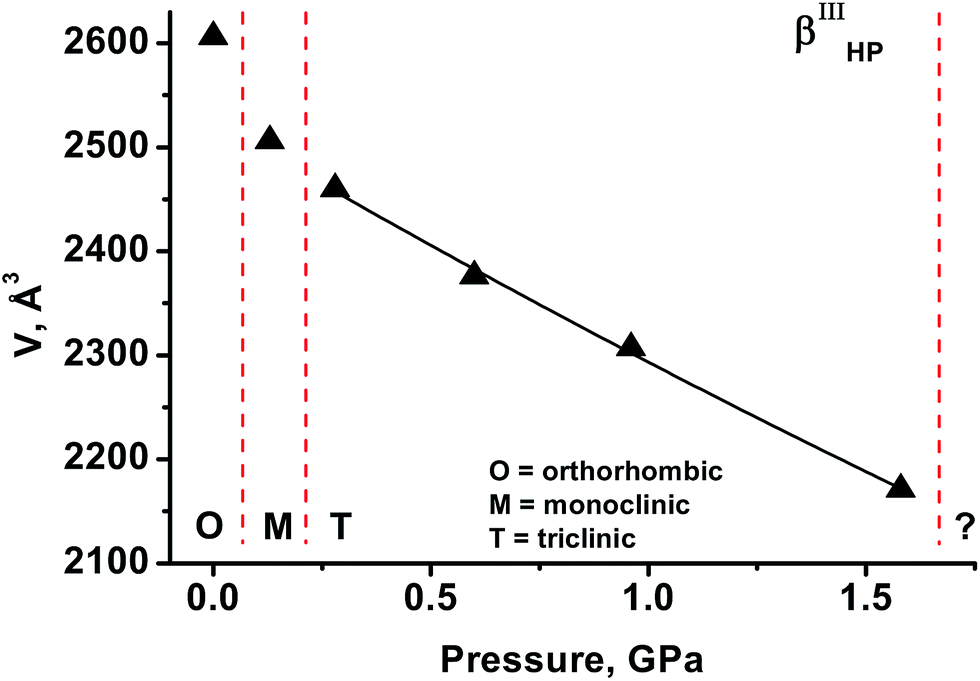 | ||
| Fig. 5 Dependency of unit cell volume on pressure for β-chlorpropamide loaded in paraffin. | ||
It has been shown that the behaviour of β-chlorpropamide on increasing pressure in several “inert” gas and liquid media is different. Helium is small enough to penetrate into the crystal structure and subsequently trigger a solid-state polymorphic transformation. The transformations observed in the presence of Ne can be expected to be of a solid-to-solid type and be defined solely by intracrystalline interactions. In the absence of He as a guest molecule, the solid-state transformation in Ne gives the α-polymorph below 0.6 GPa (the thermodynamically stable form at ambient conditions90), and not the high-pressure phases that are formed in He. Pentane–isopentane starts dissolving β-chlorpropamide at high pressure such that a high-pressure recrystallisation, and not a polymorphic solid-state transformation, takes place.89 The most interesting observation is that even the high-pressure transformations in paraffin and Ne are different, despite the fact that the two media neither dissolve nor penetrate the chlorpropamide crystal. The reason for this difference likely rests in different van der Waals interactions between the surface of chlorpropamide crystals and the pressure transmitting media in the two cases. In particular, the alkyl tails of chlorpropamide molecules can be supposed to interact with the paraffin molecules.†
Experimental
β-Chlorpropamide was crystallised by slow evaporation of saturated ethanol solutions prepared by dissolving a powder sample of α-chlorpropamide. The chlorpropamide polymorphs were loaded into DACs of different types, depending on the choice of the pressure medium (Table 3). Small ruby spheres were used as pressure calibrants.101,102 The gas loading of Ne and He did not require any preliminary cooling of the samples in the DACs. He or Ne was loaded into the DAC as a PTM using a high pressure gas loading apparatus in BGI.103 Pressure was further increased as long as the structural integrity of the crystal and the quality of diffraction data permitted determination of cell parameters. These upper limit pressure values were different for different media (see below). All diffraction data (excluding the experiment with β-chlorpropamide compressed in helium) were collected at the Swiss-Norwegian Beamline BM01A at the European Synchrotron Radiation Facility (ESRF, Grenoble, France, experiment CH-4526). A parallel monochromatic X-ray beam (E = 17.8 keV, λ = 0.69783 Å) cropped to 200 × 200 μm2 on the sample was used. Single-crystal data were collected by a horizontal-acting ω-axis rotation with an integrated step scan of 0.5° and a counting time of 2 s per frame. A PILATUS 2M hybrid pixel detector was used with sample-to-detector distance 196 mm. The data were converted and integrated using the SNBL toolbox104 and CrysAlisPro105 software packages. X-ray diffraction data for β-chlorpropamide loaded with helium were collected at ID09A synchrotron beamline (ESRF, Grenoble, France) using a parallel monochromatic X-ray beam (E = 30 keV, λ = 0.413 Å) focused to 30 × 30 μm2 on the sample. Single-crystal data were collected by a vertical-acting ω-axis rotation with an integrated step scan of 0.5° or 1° and a counting time of 1 s per frame. A MAR555 flat-panel detector was used for recording the diffraction intensities. The sample-to-detector distance was 250 mm. The data were then processed according to106 and integrated using the CrysAlisPro105 software package. Mercury107 was used to visualise the crystal structures and calculate the size of voids. The contact surfaces were calculated using the following parameters: probe radius equal to 0.3 Å for helium and to 0.4 Å for neon, approximate grid spacing is 0.3 Å.| Medium | DAC type | Gasket and crystal details |
|---|---|---|
| Ne | BX-90 type,97 an opening cone of 80°. Culet size 600 μm | Crystal size 0.16 × 0.14 × 0.02 mm, gasket: thickness: 100 μm, hole diameter: 250 μm |
| Liquid paraffin (ROTH GmbH) | BX-90 type,97 an opening cone of 80°. Culet size 600 μm | Crystal size 0.10 × 0.05 × 0.05 mm, gasket: thickness: 85 μm, hole diameter: 300 μm |
| He | A membrane-driven DAC98 with Boehler-Almax seats,99 an opening cone of 64°. Culet size 600 μm | Crystal size 0.05 × 0.04 × 0.02 mm, stainless steel gasket: thickness: 80 μm, hole diameter: 350 μm |
Conclusions
This study has shown that the choice of pressure-transmitting fluid can be critical for the outcome of a high-pressure experiment, and not only when the fluid visibly dissolves the solid. As shown in the present contribution, recrystallisation is not the only possible mechanism through which a fluid can influence a solid-state transformation in an organic molecular solid, to justify classifying the transformation as “fluid-assisted”. Other mechanisms include the penetration of the medium into the solid, generating additional inner stress and thus triggering a structural transformation, or surface interactions that can influence the mechanical properties (macroscopic level of consideration). Alternatively, fluid molecules may induce conformational changes in molecules, and thus their rearrangement into a new structure (microscopic level of consideration). The ability to change mechanical properties of solid materials (plasticity, brittleness) through addition of fluids at the surface has long been known for metals and ionic salts (Ioffe,108 Rehbinder109 or Roscoe110 effects). The same effects can be expected also for organic molecular crystals. All the various types of solid-fluid interactions, not only the possibility of dissolution or penetration, must be taken into account in any polymorph screening experiments involving high pressure as a varied parameter. Further research of the solid-fluid interfaces in these systems, of the effect of fluids on the mechanical properties of the chlorpropamide crystals, as well structure solution of the new high-pressure phases will shed more light on the mechanisms of the solid-state transformations in the presence of a fluid in these and other organic crystals. This is an area of critical importance for many fields of chemical, materials and pharmaceutical technologies.Acknowledgements
The study was supported by a grant from RSF 14-13-00834. Assistance of Prof. V. Dmitriev, Dr D. Chernyshov, Dr A. Mikheykin with the single-crystal diffraction experiment at BM01 beamline at ESRF is acknowledged. We thank Mr A. Michalchuk for language polishing and useful comments. A. Kurnosov thanks the project “GeoMaX” funded under the Emmy-Noether Program of the German Science Foundation (MA4534/3-1).Notes and references
- High Pressure Crystallography, ed. A. Katrusiak and P. McMillan, Kluwer, Dordrecht, 2003 Search PubMed.
- High-Pressure Crystallography. From Novel Experimental Approaches to Applications in Cutting-Edge Technologies, ed. E. V. Boldyreva and P. Dera, Springer, Dordrecht, 2010 Search PubMed.
- A. Katrusiak, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 135–148 CrossRef CAS PubMed.
- D. G. Billing and A. Katrusiak, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2014, 70, 399–400 CrossRef CAS PubMed.
- E. V. Boldyreva, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 218–231 CrossRef CAS PubMed.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, S. A. Moggach, S. Parsons and C. R. Pulham, CrystEngComm, 2004, 6, 504–511 RSC.
- M. A. Neumann, J. van de Streek, F. P. A. Fabbiani, P. Hidber and O. Grassmann, Nat. Commun., 2015, 6, 7793 CrossRef CAS PubMed.
- S. A. Moggach, S. Parsons and P. A. Wood, Crystallogr. Rev., 2008, 14, 143–184 CrossRef CAS.
- I. D. H. Oswald, I. Chataigner, S. Elphick, F. P. A. Fabbiani, A. R. Lennie, J. Maddaluno, W. G. Marshall, T. J. Prior, C. R. Pulham and R. I. Smith, CrystEngComm, 2009, 11, 359–366 RSC.
- F. P. A. Fabbiani and C. R. Pulham, Chem. Soc. Rev., 2006, 35, 932–942 RSC.
- E. V. Boldyreva, T. P. Shakhtshneider, H. Ahsbahs, H. Sowa and H. Uchtmann, in J. Therm. Anal. Calorim., 2002, vol. 68, pp. 437–452 Search PubMed.
- J. Bernstein, Polymorphism in Molecular Crystals, Oxford University Press, 2007 Search PubMed.
- Polymorphism: in the Pharmaceutical Industry, ed. R. Hilfiker, Wiley-VCH Verlag GmbH & Co. KGaA, Weinheim, FRG, 2006 Search PubMed.
- E. Boldyreva, Cryst. Growth Des., 2007, 7, 1662–1668 CAS.
- E. V. Boldyreva, S. N. Ivashevskaya, H. Sowa, H. Ahsbahs and H. P. Weber, Dokl. Akad. Nauk, 2004, 396, 358–361 Search PubMed.
- E. V. Boldyreva, S. N. Ivashevskaya and H. Sowa, Z. Kristallogr. Cryst. Mater., 2005, 220, 50–57 CAS.
- S. V. Goryainov, E. N. Kolesnik and E. V. Boldyreva, Phys. B, 2005, 357, 340–347 CrossRef CAS.
- N. A. Tumanov, E. V. Boldyreva and H. Ahsbahs, Powder Diffr., 2008, 23, 307–316 CrossRef CAS.
- S. V. Goryainov, E. V. Boldyreva and E. N. Kolesnik, Chem. Phys. Lett., 2006, 419, 496–500 CrossRef CAS.
- J. Ridout, L. S. Price, J. A. K. Howard and M. R. Probert, Cryst. Growth Des., 2014, 14, 3384–3391 CAS.
- B. A. Zakharov, N. A. Tumanov and E. V. Boldyreva, CrystEngComm, 2015, 17, 2074–2079 RSC.
- M. Fisch, A. Lanza, E. Boldyreva, P. Macchi and N. Casati, J. Phys. Chem. C, 2015, 119, 18611–18617 CAS.
- N. A. Tumanov, E. V. Boldyreva, B. A. Kolesov, A. V. Kurnosov and R. Quesada Cabrera, Acta Crystallogr., Sect. B: Struct. Sci., 2010, 66, 458–471 CAS.
- E. V. Boldyreva, H. Ahsbahs, H. Uchtmann and N. Kashcheeva, High Pressure Res., 2000, 17, 79–99 CrossRef.
- E. V. Boldyreva, V. Dmitriev and B. C. Hancock, Int. J. Pharm., 2006, 327, 51–57 CrossRef CAS PubMed.
- Y. V. Seryotkin, T. N. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2013, 69, 77–85 CAS.
- E. Eikeland, M. K. Thomsen, S. R. Madsen, J. Overgaard, M. A. Spackman and B. B. Iversen, Chem.–Eur. J., 2016, 22, 4061–4069 CrossRef CAS PubMed.
- W. Zielinski and A. Katrusiak, CrystEngComm, 2016, 18, 3211–3215 RSC.
- G. Gottardi and E. Galli, Natural zeolites, Springer-Verlag, Berlin, 1985 Search PubMed.
- I. A. Belitsky, B. A. Fursenko, S. P. Gabuda, O. V. Kholdeev and Y. V. Seryotkin, Phys. Chem. Miner., 1992, 18, 497–505 CrossRef.
- Y. Lee, J. A. Hriljac, T. Vogt, J. B. Parise and G. Artioli, J. Am. Chem. Soc., 2001, 123, 12732–12733 CrossRef CAS PubMed.
- Y. V. Seryotkin, V. V. Bakakin, B. A. Fursenko, I. A. Belitsky, W. Joswig and P. G. Radaelli, Eur. J. Mineral., 2005, 17, 305–314 CrossRef CAS.
- S. V. Rashchenko, Y. V. Seryotkin and V. V. Bakakin, Microporous Mesoporous Mater., 2012, 159, 126–131 CrossRef CAS.
- S. Quartieri, G. Montagna, R. Arletti and G. Vezzalini, J. Solid State Chem., 2011, 184, 1505–1516 CrossRef CAS.
- Y. V. Seryotkin, Microporous Mesoporous Mater., 2015, 214, 127–135 CrossRef CAS.
- G. D. Gatta and Y. Lee, Microporous Mesoporous Mater., 2007, 105, 239–250 CrossRef CAS.
- Y. V. Seryotkin, Microporous Mesoporous Mater., 2016, 226, 415–423 CrossRef CAS.
- G. Shen, Q. Mei, V. B. Prakapenka, P. Lazor, S. Sinogeikin, Y. Meng and C. Park, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 6004–6007 CrossRef CAS PubMed.
- B. Coasne, C. Weigel, A. Polian, M. Kint, J. Rouquette, J. Haines, M. Foret, R. Vacher and B. Rufflé, J. Phys. Chem. B, 2014, 118, 14519–14525 CrossRef CAS PubMed.
- K. Niwa, T. Tanaka, M. Hasegawa, T. Okada, T. Yagi and T. Kikegawa, Microporous Mesoporous Mater., 2013, 182, 191–197 CrossRef CAS.
- T. Sato, N. Funamori and T. Yagi, Nat. Commun., 2011, 2, 345 CrossRef PubMed.
- T. Sato, H. Takada, T. Yagi, H. Gotou, T. Okada, D. Wakabayashi and N. Funamori, Phys. Chem. Miner., 2013, 40, 3–10 CrossRef CAS.
- C. Weigel, A. Polian, M. Kint, B. Rufflé, M. Foret and R. Vacher, Phys. Rev. Lett., 2012, 109, 245504 CrossRef PubMed.
- B. A. Zakharov and A. F. Achkasov, J. Appl. Crystallogr., 2013, 46, 267–269 CAS.
- R. J. Angel, M. Bujak, J. Zhao, G. D. Gatta and S. D. Jacobsen, J. Appl. Crystallogr., 2007, 40, 26–32 CrossRef CAS.
- S. Klotz, J.-C. Chervin, P. Munsch and G. Le Marchand, J. Phys. D: Appl. Phys., 2009, 42, 075413 CrossRef.
- W. Cai, A. Gładysiak, M. Anioła, V. J. Smith, L. J. Barbour and A. Katrusiak, J. Am. Chem. Soc., 2015, 137, 9296–9301 CrossRef CAS PubMed.
- A. J. Graham, A.-M. Banu, T. Düren, A. Greenaway, S. C. McKellar, J. P. S. Mowat, K. Ward, P. A. Wright and S. A. Moggach, J. Am. Chem. Soc., 2014, 136, 8606–8613 CrossRef CAS PubMed.
- J. Joo, H. Kim and S. S. Han, Phys. Chem. Chem. Phys., 2013, 15, 18822–18826 RSC.
- C. L. Hobday, R. J. Marshall, C. F. Murphie, J. Sotelo, T. Richards, D. R. Allan, T. Düren, F.-X. Coudert, R. S. Forgan, C. A. Morrison, S. A. Moggach and T. D. Bennett, Angew. Chem., Int. Ed., 2016, 55, 2401–2405 CrossRef CAS PubMed.
- S. C. McKellar, J. Sotelo, A. Greenaway, J. P. S. Mowat, O. Kvam, C. A. Morrison, P. A. Wright and S. A. Moggach, Chem. Mater., 2016, 28, 466–473 CrossRef CAS.
- S. C. McKellar, J. Sotelo, J. P. S. Mowat, P. A. Wright and S. A. Moggach, CrystEngComm, 2016, 18, 1273–1276 RSC.
- S. C. McKellar and S. A. Moggach, Acta Crystallogr., Sect. B: Struct. Sci., Cryst. Eng. Mater., 2015, 71, 587–607 CrossRef CAS PubMed.
- S. C. McKellar, A. J. Graham, D. R. Allan, M. I. H. Mohideen, R. E. Morris and S. A. Moggach, Nanoscale, 2014, 6, 4163–4173 RSC.
- W. Cai and A. Katrusiak, Nat. Commun., 2014, 5, 4337 CAS.
- F. P. A. Fabbiani, S. Bergantin, A. Gavezzotti, S. Rizzato and M. Moret, CrystEngComm, 2016, 18, 2173–2181 RSC.
- F. P. A. Fabbiani, G. Buth, D. C. Levendis and A. J. Cruz-Cabeza, Chem. Commun., 2014, 50, 1817–1819 RSC.
- R. Granero-García, F. J. Lahoz, C. Paulmann, S. Saouane and F. P. A. Fabbiani, CrystEngComm, 2012, 14, 8664–8670 RSC.
- F. P. A. Fabbiani, D. C. Levendis, G. Buth, W. F. Kuhs, N. Shankland and H. Sowa, CrystEngComm, 2010, 12, 2354–2360 RSC.
- F. P. A. Fabbiani, B. Dittrich, A. J. Florence, T. Gelbrich, M. B. Hursthouse, W. F. Kuhs, N. Shankland and H. Sowa, CrystEngComm, 2009, 11, 1396–1406 RSC.
- F. P. A. Fabbiani, D. R. Allan, W. I. F. David, A. J. Davidson, A. R. Lennie, S. Parsons, C. R. Pulham and J. E. Warren, Cryst. Growth Des., 2007, 7, 1115–1124 CAS.
- F. P. A. Fabbiani, D. R. Allan, S. Parsons and C. R. Pulham, CrystEngComm, 2005, 7, 179–186 RSC.
- F. P. A. Fabbiani, D. R. Allan, W. G. Marshall, S. Parsons, C. R. Pulham and R. I. Smith, J. Cryst. Growth, 2005, 275, 185–192 CrossRef CAS.
- F. P. A. Fabbiani, D. R. Allan, A. Dawson, W. I. F. David, P. A. McGregor, I. D. H. Oswald, S. Parsons and C. R. Pulham, Chem. Commun., 2003, 9, 3004–3005 RSC.
- A. Olejniczak, M. Podsiadło and A. Katrusiak, New J. Chem., 2016, 40, 2014–2020 RSC.
- W. Zieliński and A. Katrusiak, CrystEngComm, 2015, 17, 5468–5473 RSC.
- M. Anioła, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2014, 14, 2187–2191 Search PubMed.
- H. Tomkowiak, A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2013, 13, 121–125 CAS.
- A. Olejniczak and A. Katrusiak, Cryst. Growth Des., 2011, 11, 2250–2256 CAS.
- A. Olejniczak and A. Katrusiak, CrystEngComm, 2010, 12, 2528–2532 RSC.
- Y. Sakata, Agric. Biol. Chem., 1961, 25, 829–837 CAS.
- E. Sheikholeslamzadeh and S. Rohani, Ind. Eng. Chem. Res., 2013, 52, 2633–2641 CrossRef CAS.
- J. Schöll, D. Bonalumi, L. Vicum, M. Mazzotti and M. Müller, Cryst. Growth Des., 2006, 6, 881–891 Search PubMed.
- E. V. Boldyreva, S. G. Arkhipov, T. N. Drebushchak, V. A. Drebushchak, E. A. Losev, A. A. Matvienko, V. S. Minkov, D. A. Rychkov, Y. V. Seryotkin, J. Stare and B. A. Zakharov, Chem.–Eur. J., 2015, 21, 15395–15404 CrossRef CAS PubMed.
- E. V. Boldyreva, V. A. Drebushchak, I. E. Paukov, Y. A. Kovalevskaya and T. N. Drebushchak, J. Therm. Anal. Calorim., 2004, 77, 607–623 CrossRef CAS.
- C. Näther, I. Jess, P. Kuś and P. G. Jones, CrystEngComm, 2016, 18, 3142–3149 RSC.
- C. Iwaihara, D. Kitagawa and S. Kobatake, Cryst. Growth Des., 2015, 15, 2017–2023 CAS.
- L. Wang, Y. Song, P. Yang, B. Tan, H. Zhang and Z. Deng, J. Therm. Anal. Calorim., 2015, 120, 947–951 CrossRef CAS.
- R. Bobrovs, L. Seton and A. Actiņš, CrystEngComm, 2014, 16, 10581–10591 RSC.
- C. Näther, I. Jess, J. Bahrenburg, D. Bank and F. Temps, CrystEngComm, 2014, 16, 5633–5641 RSC.
- W. Du, Q. Yin, H. Hao, Y. Bao, X. Zhang, J. Huang, X. Li, C. Xie and J. Gong, Ind. Eng. Chem. Res., 2014, 53, 5652–5659 CrossRef CAS.
- K. Kawakami, Y. Asami and I. Takenoshita, J. Pharm. Sci., 2010, 99, 76–81 CrossRef CAS PubMed.
- R. Arletti, L. Leardini, G. Vezzalini, S. Quartieri, L. Gigli, M. Santoro, J. Haines, J. Rouquette and L. Konczewicz, Phys. Chem. Chem. Phys., 2015, 17, 24262–24274 RSC.
- A. P. Ayala, M. W. C. Caetano, S. B. Honorato, J. Mendes Filho, H. W. Siesler, S. N. Faudone, S. L. Cuffini, F. T. Martins, C. C. P. da Silva and J. Ellena, J. Raman Spectrosc., 2012, 43, 263–272 CrossRef CAS.
- A. J. Cruz-Cabeza and J. Bernstein, Chem. Rev., 2014, 114, 2170–2191 CrossRef CAS PubMed.
- J. Wąsicki, D. P. Kozlenko, S. E. Pankov, P. Bilski, A. Pajzderska, B. C. Hancock, A. Medek, W. Nawrocik and B. N. Savenko, J. Pharm. Sci., 2009, 98, 1426–1437 CrossRef PubMed.
- S. E. Kichanov, D. P. Kozlenko, J. Wąsicki, W. Nawrocik, L. S. Dubrovinsky, H.-P. Liermann, W. Morgenroth and B. N. Savenko, J. Pharm. Sci., 2015, 104, 81–86 CrossRef CAS PubMed.
- N. V. Loshak, S. E. Kichanov, D. P. Kozlenko, J. Wąsicki, W. Nawrocik, E. V. Lukin, C. Lathe, B. N. Savenko and L. A. Bulavin, J. Surf. Invest.: X-Ray, Synchrotron Neutron Tech., 2012, 6, 951–953 CrossRef CAS.
- B. A. Zakharov, S. V. Goryainov and E. V. Boldyreva, CrystEngComm, 2016, 18, 5423–5428 RSC.
- V. A. Drebushchak, T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, J. Therm. Anal. Calorim., 2008, 93, 343–351 CrossRef CAS.
- T. N. Drebushchak, N. V. Chukanov and E. V. Boldyreva, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2006, 62, o4393–o4395 CAS.
- P. A. Guńka, K. F. Dziubek, A. Gładysiak, M. Dranka, J. Piechota, M. Hanfland, A. Katrusiak and J. Zachara, Cryst. Growth Des., 2015, 15, 3740–3745 Search PubMed.
- R. T. Downs and D. C. Palmer, Am. Mineral., 1994, 79, 9–14 CAS.
- T. N. Drebushchak, V. A. Drebushchak and E. V. Boldyreva, Acta Crystallogr., Sect. B: Struct. Sci., 2011, 67, 163–176 CAS.
- J. A. Sans, F. J. Manjón, C. Popescu, V. P. Cuenca-Gotor, O. Gomis, A. Muñoz, P. Rodríguez-Hernández, J. Contreras-García, J. Pellicer-Porres, A. L. J. Pereira, D. Santamaría-Pérez and A. Segura, Phys. Rev. B, 2016, 93, 054102 CrossRef.
- C. Lobban, J. L. Finney and W. F. Kuhs, J. Chem. Phys., 2002, 117, 3928–3934 CrossRef CAS.
- I. Kantor, V. Prakapenka, A. Kantor, P. Dera, A. Kurnosov, S. Sinogeikin, N. Dubrovinskaia and L. Dubrovinsky, Rev. Sci. Instrum., 2012, 83, 125102 CrossRef CAS PubMed.
- R. Letoullec, J. P. Pinceaux and P. Loubeyre, High Pressure Res., 1988, 1, 77–90 CrossRef.
- R. Boehler and K. De Hantsetters, High Pressure Res., 2004, 24, 391–396 CrossRef.
- T. N. Drebushchak, A. A. Ogienko and E. V. Boldyreva, CrystEngComm, 2011, 13, 4405–4410 RSC.
- R. A. Forman, G. J. Piermarini, J. Dean Barnett and S. Block, Science, 1972, 176, 284–285 CAS.
- G. J. Piermarini, S. Block, J. D. Barnett and R. A. Forman, J. Appl. Phys., 1975, 46, 2774–2780 CrossRef CAS.
- A. Kurnosov, I. Kantor, T. Boffa-Ballaran, S. Lindhardt, L. Dubrovinsky, A. Kuznetsov and B. H. Zehnder, Rev. Sci. Instrum., 2008, 79, 045110 CrossRef PubMed.
- V. Dyadkin, SNBL Tool box, Swiss-Norwegian Beamlines at ESRF, Grenoble, France, 2013 Search PubMed.
- CrysAlisPro, Agilent Technologies UK, Yarnton, Oxfordshire, UK, 2013 Search PubMed.
- A. Rothkirch, G. D. Gatta, M. Meyer, S. Merkel, M. Merlini and H.-P. Liermann, J. Synchrotron Radiat., 2013, 20, 711–720 CAS.
- C. F. Macrae, P. R. Edgington, P. McCabe, E. Pidcock, G. P. Shields, R. Taylor, M. Towler and J. van de Streek, J. Appl. Crystallogr., 2006, 39, 453–457 CrossRef CAS.
- A. F. Ioffe, Usp. Fiz. Nauk, 1936, 16, 847–871 CrossRef.
- P. Rehbinder, Nature, 1947, 159, 866–867 CrossRef.
- R. Roscoe, Nature, 1934, 133, 912 CrossRef.
Footnote |
| † It has been shown previously that ball-milling of α-chlorpropamide results in a polymorphic transformation into the ε-polymorph only at 77 K, but not at ambient temperature. It has been supposed, that a change in the orientation of the alkyl tails in chlorpropamide molecules on cooling can trigger a polymorphic transformation on milling at low temperatures.100 |
| This journal is © The Royal Society of Chemistry 2016 |