
Re-evaluation of the N-terminal substitution and the D-residues of teixobactin†
Shimaa A. H. Abdel Monaima,
Yahya E. Jada,
Gerardo A. Acostab,
Tricia Naickera,
Estelle J. Ramchurana,
Ayman El-Fahamcd,
Thavendran Govendera,
Hendrik G. Krugera,
Beatriz G. de la Torrea and
Fernando Albericio *abdef
*abdef
aCatalysis and Peptide Research Unit, School of Health Sciences, University of KwaZulu-Natal, Durban 4001, South Africa. E-mail: albericio@ukzn.ac.za
bCIBER-BBN, Networking Centre on Bioengineering, Biomaterials and Nanomedicine, Barcelona Science Park, 08028-Barcelona, Spain
cDepartment of Chemistry, Faculty of Science, Alexandria University, P.O. Box 426, Ibrahimia, Alexandria 21321, Egypt
dDepartment of Chemistry, College of Science, King Saud University, P.O. Box 2455, Riyadh 11451, Saudi Arabia
eSchool of Chemistry and Physics, University of KwaZulu-Natal, Durban 4001, South Africa
fDepartment of Organic Chemistry, University of Barcelona, 08028-Barcelona, Spain
First published on 28th July 2016
Abstract
Teixobactin is a head to side-chain cyclic depsipeptide with a guanidino based residue within the cycle, three D-amino acids in the tail, and a N-methylated terminal residue. The synthesis of the first analogue, containing Arg, was recently described by our group. Herein, we demonstrated that analogues of Arg. Teixobactin bearing either (a) three L-amino acids in the tail and keeping the N-methyl at the N-terminal or (b) with three D-amino acids, but with acetylation of the N-terminal, are inactive against Gram(+) and Gram(−) bacteria. These results complement those published by the groups of Madder, Taylor, and Singh that have shown that both modifications: L-amino acids and N-acetylation also led to loss of biological activity.
The development of new antimicrobial drugs has become essential and crucial due the dramatic increase of bacterial resistance to traditional antibiotics.1–3 Amongst these new antimicrobial compounds, antimicrobial peptides (AMP), especially cyclic peptides, have been shown to be very appealing due to the potent activities and stability.4–6 Very recently (2015), a new head to side chain antimicrobial cyclodepsipetides called teixobactin (1) was discovered by Ling et al. using an iChip multichannel device as a new discovery technique to simultaneously isolate and grow uncultured bacteria.7 This new antimicrobial peptide exhibited higher activity against Gram positive than Gram negative bacteria. Furthermore, teixobactin has been reported to kill bacteria in absence of any detectable resistance. The reason for that was ascribed to its mechanism of action; where it blocks one of the membrane-associated steps of peptidoglycan biosynthesis and then causes inhibition of lipid II, as a predominant target, and lipid III which plays a critical role in the synthesis of teichoic acid.7,8
Since the publication of isolation, characterization, and antimicrobial activity of teixobactin,7 several groups started the development of synthetic strategies for a convenient synthesis that is suitable for running a medicinal chemistry program. Our group published in 2015, the first synthesis of an analogue of teixobactin (2a) in 2015 where the non-proteinogenic guanidine-based amino acid enduracididine was substituted with the proteinogenic arginine.9 This analogue showed similar biological trends, being active against the Gram-positive bacteria and inactive against Gram-negative bacteria. However, teixobcatin was one order of magnitude more active than the Arg analogue.9,10
At the beginning of 2016, the groups of Madder, Taylor, and Singh published the synthesis of the same analogue, Arg–teixobactin (2a),11 using an identical strategy and reporting the same biological activity. In the same manuscript, the authors stated that “(they) established that the D-amino acids are critical for the antimicrobial activity”. They supported this statement with the synthesis of a second analogue (3b) where the three D-amino acids of the tail were substituted by the L-amino acid residues (Phe, Gln, Ile). However, the N-Me of the terminal Phe for this new analogue (3b) was substituted with N-Ac. Substitution of N-Me-Phe residue with Ac-Phe should have a significant impact in the biological activity. The impact of this Ac-Phe terminal could potentially be more critical than the change to L-amino acids, since the peptide (3b) has lost its cationic character (a normal requirement for antimicrobial peptides)12 due to introduction of the acetyl group. In addition to that, the hydrogen bond map of the peptide is also dramatically altered.
Herein, we report the synthesis and biological evaluation of two unpublished analogues: (2b) that has three L-amino acids while keeping the N-Me group at the Phe terminal; and (3a) with D-amino acids, but where the D-Phe terminal is acetylated (Fig. 1).
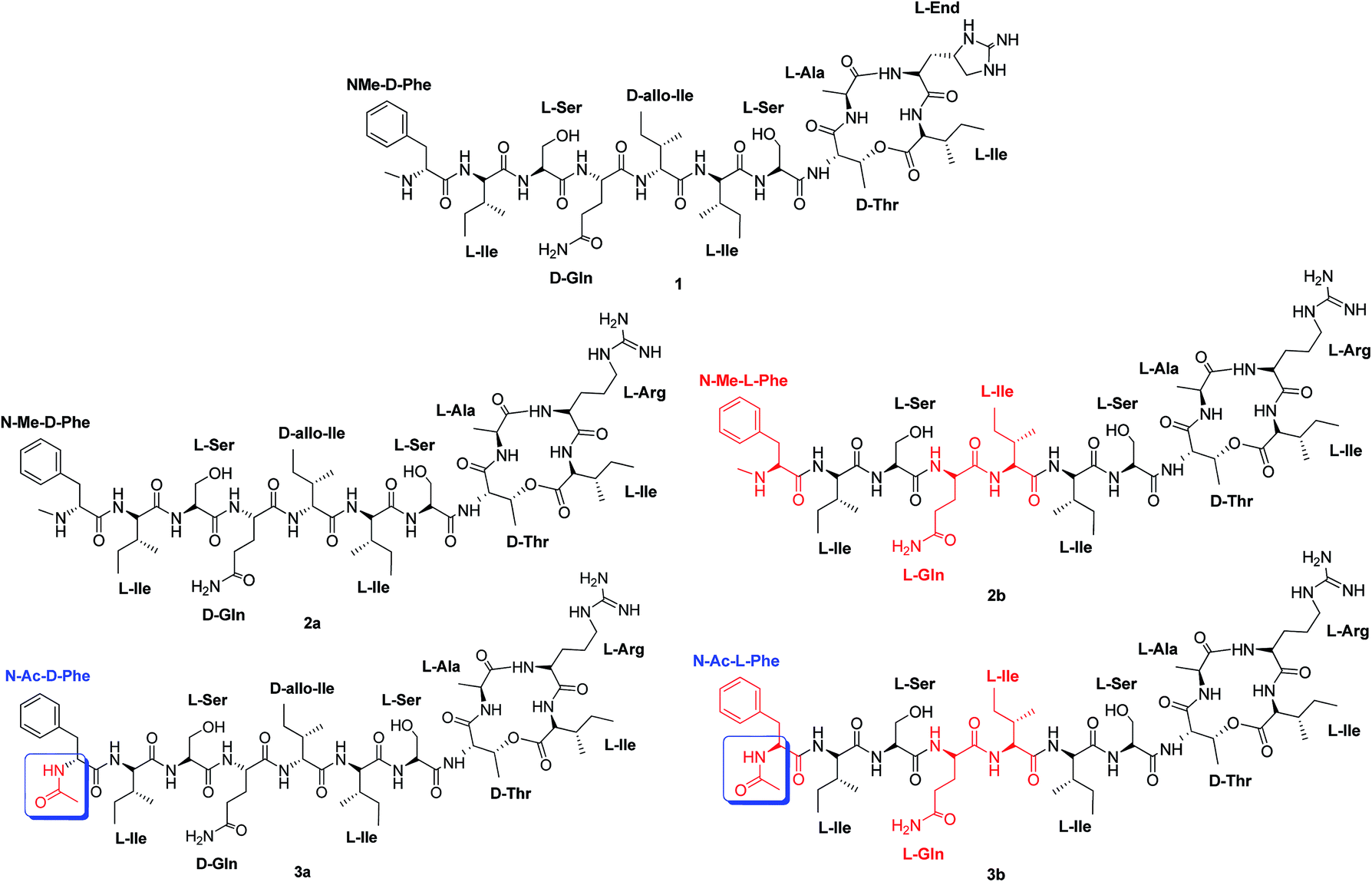 | ||
| Fig. 1 Structure of teixobactin. (1) Structure of teixobactin analogue (2a) with D-amino acids at the tail and a N-(Me)-terminal.6,9 Structure of teixobactin analogue (2b) with L-amino acids at the tail and a N-(Me)-terminal (new analogue). Structure of teixobactin analogue (3a) with D-amino acids at the tail and a N-(Ac)-terminal (new analogue). Structure of teixobactin analogue (3b) with L-amino acids at the tail and a N-(Ac)-terminal.11 | ||
Syntheses of analogues (2b) and (3b) were carried following basically the same synthetic scheme used by our group for the synthesis of Arg–teixobactin (2a),9 (Schemes 1 and 2), with the only modification for the analogue (3a) where Fmoc protection was used for the D-Phe, instead of Boc. Before cleavage of the latter from the resin, the Fmoc group was removed and the free amine was acetylated with HOAc in presence of K-Oxyma/DIC. Both analogues were obtained with an overall non-optimized yield of 50% for (2b) and 56% for (3a) before purification. The respective yields after purification were 3.8% for (2b) and 3.9% for (3a), and these peptides were characterized by HPLC and HRMS.
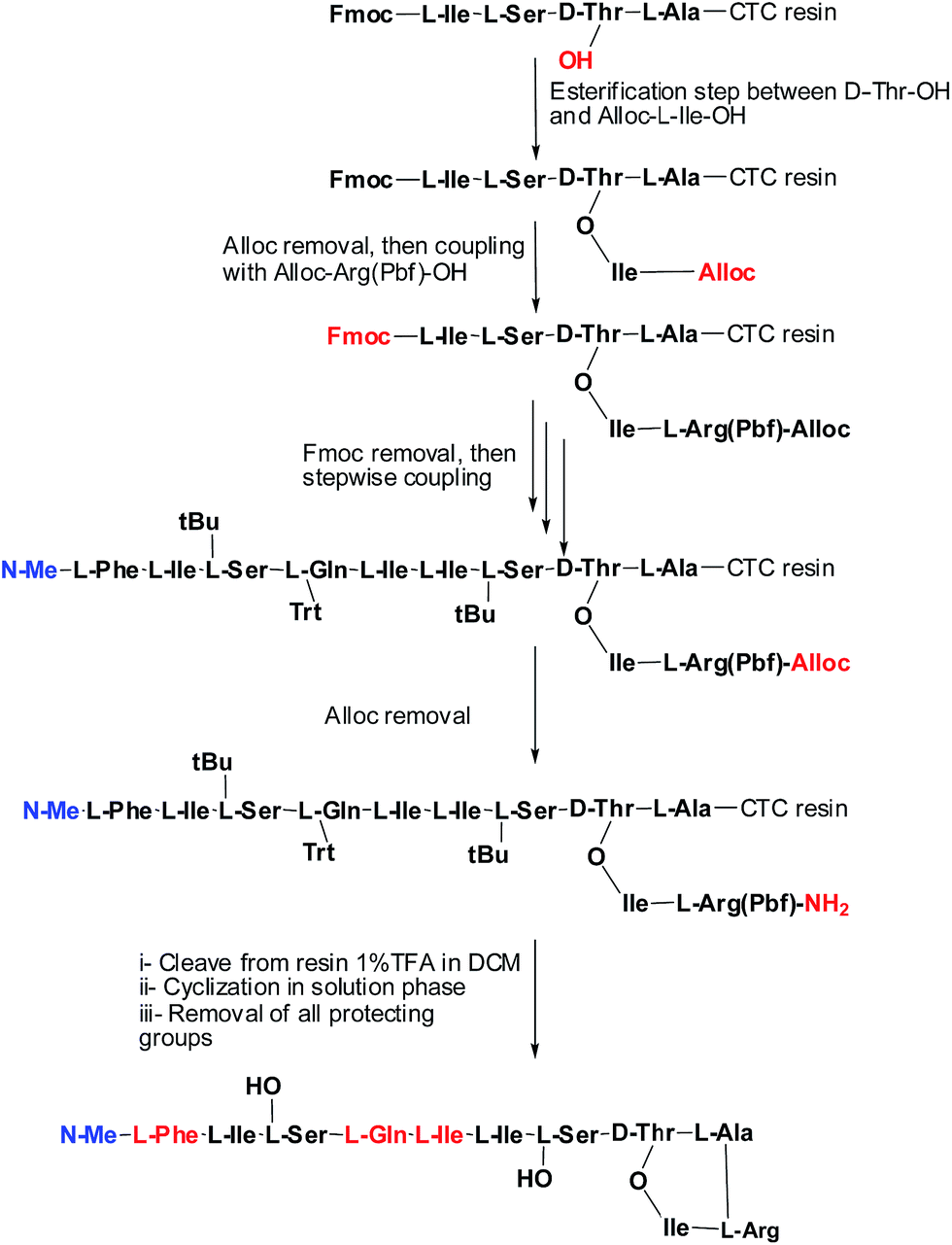 | ||
| Scheme 1 Synthetic strategy of (2b). | ||
 | ||
| Scheme 2 Synthetic strategy of (3a). | ||
Analogues (2b) and (3a) were evaluated against two ATCC Gram positive bacterial strains (Staphylococcus aureus 29213 and Bacillus subtilis 6051) and two ATCC Gram negative bacterial strains (Escherichia coli 25822 and Pseudomonas aeruginosa 27853) resulting in a total lack of activity as shown in (Table 1).
| Entry | MIC μg ml−1 (ATCC strains) | |||
|---|---|---|---|---|
| Gram(+) | Gram(−) | |||
| S. aureus | B. subtilis | E. coli | P. aeruginosa | |
| a NI-no inhibition.b GAW: growth in all wells.c Use as control in our experiments. | ||||
| Teixobactin7 | 0.25 | 0.06 | 25 | >32 |
| 2a9 | 1.99 | 0.49 | 63.9 | NI |
| 2a11 | 2 | — | 64 | — |
| 2b | NIa | NI | NI | NI |
| 3a | NI | 256 | NI | NI |
| 3b11 | 128 | — | GAWb | — |
| Meropenemc | 0.0625 | 0.0625 | 0.03125 | 0.25 |
In conclusion, the synthesis of two new analogues of 2a was achieved. Peptide 2b is different to 2a in that three D-amino acids have been substituted by the L-residues (Phe, Gln, Ile), while 3a is different to the teixobactin analogue 2a only in that the terminal N-Me group was replaced with a N-Ac group. Both of these compounds lost totally the bacterial activity. These results complement those reported by the groups of Madder, Taylor, and Singh that have found that the double change (3b), L-amino acids and acetyl at the N-terminal acetylated is also inactive.
Acknowledgements
This work was funded in part by the following: the National Research Foundation (NRF) and the University of KwaZulu-Natal (South Africa); the CICYT (CTQ2012-30930), and the Generalitat de Catalunya (2014 SGR 137).Notes and references
- L. H. Kondejewski, M. Jelokhani-Niaraki, S. W. Farmer, B. Lix, C. M. Kay, B. D. Sykes, R. E. W. Hancock and R. S. Hodges, J. Biol. Chem., 1999, 274, 13181–13192 CrossRef CAS PubMed.
- H. C. Neu, Science, 1992, 257, 1064–1073 CAS.
- C. M. Kunin, Ann. Intern. Med., 1993, 118, 557–561 CrossRef CAS PubMed.
- S. Fernandez-Lopez, H.-S. Kim, E. C. Choi, M. Delgado, J. R. Granja, A. Khasanov, K. Kraehenbuehl, G. Long, D. A. Weinberger and K. M. Wilcoxen, Nature, 2001, 412, 452–455 CrossRef CAS PubMed.
- P. Thapa, M. J. Espiritu, C. Cabalteja and J.-P. Bingham, Int. J. Pept. Res. Ther., 2014, 20, 545–551 CrossRef CAS.
- S.-H. Joo, Biomol. Ther., 2012, 20, 2 Search PubMed.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schäberle, D. E. Hughes and S. Epstein, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- A. Penesyan, M. Gillings and I. T. Paulsen, Molecules, 2015, 20, 5286–5298 CrossRef CAS PubMed.
- Y. E. Jad, G. A. Acosta, T. Naicker, M. Ramtahal, A. El-Faham, T. Govender, H. G. Kruger, B. G. D. L. Torre and F. Albericio, Org. Lett., 2015, 17, 6182–6185 CrossRef CAS PubMed.
- During the preparation of this manuscript, it has appeared published the first total synthesis of teixobactin: A. M. Giltrap, L. J. Dowman, G. Nagalingam, J. L. Ochoa, R. G. Linington, W. J. Britton and R. J. Payne, Org. Lett., 2016, 18, 2788–2791 CrossRef CAS PubMed.
- A. Parmar, A. Iyer, C. S. Vincent, D. Van Lysebetten, S. H. Prior, A. Madder, E. J. Taylor and I. Singh, Chem. Commun., 2016, 52, 6060–6063 RSC.
- M. Wu and R. E. Hancock, J. Biol. Chem., 1999, 274, 29–35 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic method, HPLC, HRMS and MALDI. See DOI: 10.1039/c6ra17720d |
| This journal is © The Royal Society of Chemistry 2016 |