DOI:
10.1039/C6RA17690A
(Paper)
RSC Adv., 2016,
6, 88179-88188
Double-channel emission from gold nanoparticles functionalized with a thermo-responsive copolymer ligand: preparation, optical properties and control of catalytic activity†
Received
11th July 2016
, Accepted 31st August 2016
First published on 31st August 2016
Abstract
Multifunctional fluorescent gold nanoparticles (Au NPs) were obtained in situ with the assistance of a novel thermo-responsive copolymer, p(NIPAM-co-ETMA) (PNE), containing NIPAM units and an episulfide moiety, for the first time. The PNE-decorated Au NPs can emit multiple colors due to the double-channel in their intrinsic luminescence core (460 nm) and electron transfer between the PNE ligand and unreduced Au+ on the surface of the Au NPs (530 nm). Moreover, these NPs capped with Au+ on the surface were able to efficiently catalyze the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) by NaBH4 in an aqueous environment. The catalytic activity can be controlled by thermo-switching due to the sensitive thermally triggered response of PNE.
Introduction
The development of metallic nanostructures has been the subject of intensive research over several decades due to its fundamental scientific interest and the importance of this technology in a variety of applications.1–5 Especially, gold nanoparticles (Au NPs), which have attracted much attention due to their small size, may have markedly different properties from those of the respective bulk metals, such as special optoelectronic properties, fluorescence properties, and plasma resonance.6 Few-atom gold nanoclusters (Au NCs) have attracted great interest because they bridge the evolution of properties from isolated atoms to nanoparticles, and even to the bulk.7 Au NPs are attractive and potentially useful for bioimaging and biosensing in live cells due to their excellent features, including long lifetime, comparable size to biopolymers, water solubility, and biocompatibility.8 Au NPs also have intriguing molecular-like properties, such as a discrete electronic state, strong luminescence, ultra-small size, excellent photo stability, and high sensitivity to the environment.9 The emission center of gold nanoclusters is usually assigned to the inter-band transition, Au 5d10 to 6sp and/or the ligand–metal charge transfer transition.10 Wu and Jin11 found that ligands with electron-rich atoms (N, O) or groups (COOH, NH2) had a significant influence on the fluorescence properties of gold nanoclusters. In addition to the special optical, electrical, and fluorescence properties, the catalytic activity of gold nanoparticles or nanoclusters has also been a source of interest. For example, Jin et al. reported the high catalytic activity of ultra-small Au NPs toward the semihydrogenation of terminal alkynes into alkenes with high yield.12 In these areas, one of the main problems of the nanoparticles is their limited colloidal stability, which results in a tendency to form aggregates in aqueous media due to their highly active surface atoms, and subsequently leads to reduced catalytic activity. In order to improve the colloidal stability of Au NPs, the preparation of metal nanoparticles is usually carried out by the reduction of metal ions in the presence of a stabilizing polymer.13 There are different ways to connect the polymer to the surface of nanoparticles. The classical method is in situ reduction techniques, and this strategy is simple and convenient.14 Because thiol ligands have a strong affinity for gold, they are also the most commonly used in the preparation of gold nanoparticles or gold nanoclusters.15 And sulfur-containing non-thiol ligands can also stabilize gold nanoparticles. Rao et al. used the ability of lipoic acid (α-LA) to chelate metal ions for the in situ reduction of gold nanoparticles.16 In addition, dendrimers,17 DNA,18 and proteins19 can also be used as effective ligands to protect gold nanoparticles (and nanoclusters). Multifunctional nanoparticles combine multiple functions in one nanoparticle, and have received much attention. For example, by combining the two functions of catalysis and thermal sensitivity in nanoparticles, researchers can effectively control the rate of a catalytic reaction by adjusting the temperature.20 Poly(N-isopropylacrylamide) (PNIPAM) is the most popularly studied thermo-responsive polymer,21 and can undergo a reversible “coil-to-globule” phase transition in water around its lower critical solution temperature (LCST), due to a fine hydrophobic–hydrophilic balance in its structure.22,23 Zhang et al. loaded gold nanoparticles into a micelle formed by the self-assembly of the block copolymer, PNIPAM-b-P4VP, and the on/off catalysis of gold particles could be realized based on the thermal properties of PNIPAM.24
It is well known that thiols are widely employed as anchoring groups via Au-to-thiol bonds. However, with theoretical analysis, it is very easy to form two sulfur bonds that may enhance the crosslinking of the polymers. Episulfide compounds are able to chelate heavy metal ions via ring-opening reactions under acidic or basic conditions.25 The episulfide monomer of 2,3-epithiopropyl methacrylate (ETMA) as a ligand is very stable and has a strong coordination ability with metal. Herein, we reported a new type of episulfide-group-containing thermo-responsive copolymers, which can be used as effective ligands for the synthesis of Au NPs.
In this paper, the novel random copolymer ligands p(NIPAM-co-ETMA) (PNE), composed of NIPAM and the episulfide monomer of ETMA, were synthesized via conventional free-radical copolymerization (Scheme 1). Then, multifunctional Au NPs, which combine photoluminescence and catalytic activity, were prepared through the in situ reduction of an Au3+ complex with NaBH4, or alkali in the presence of PNE (Scheme 2), since the strongly acidic HAuCl4 can induce episulfide-group ring-opening to form an Au3+-S-PNE complex. The fluorescent properties and temperature-dependent catalytic activity of PNE-functionalized Au NPs were studied and the mechanism of double-channel emission by Au NPs was further proved by an NaBH4 reduction experiment and X-ray photoelectron spectroscopy (XPS). Moreover, the observed temperature-dependent catalytic behavior of Au@NPs can make them potentially important for controlling catalytic activity and catalyst recycling.
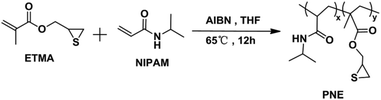 |
| | Scheme 1 General scheme for the synthesis of p(NIPAM-co-ETMA) copolymer. | |
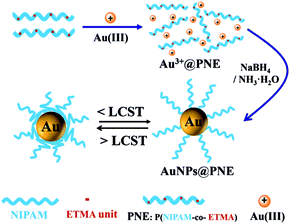 |
| | Scheme 2 Schematic of the structure of the copolymer and Au NPs@PNE formed by in situ reduction in an aqueous solution. | |
Experimental
Materials
Gold(III) chloride trihydrate (HAuCl4·3H2O, 99%), 4-nitrophenol (4-NP, 99%), and sodium borohydride (NaBH4, 96%) were obtained from Sinopharm Chemical Reagent Co. Ltd. Glycidyl methacrylate (GMA, 99.44%) was purchased from Dow Chemical Co. N-Isopropylacrylamide (NIPAM, Aldrich) was recrystallized in hexane; 2,2′-azobis(2-methylpropionitrile) (AIBN) (98%, Aldrich) was recrystallized twice from ethanol and stored in the dark at 4 °C. Tetrahydrofuran (THF) was purified by refluxing over sodium wire and distilled prior to use. Other chemicals were of analytical grade and were used as received.
Preparation of p(NIPAM-co-ETMA) (PNE)
The monomer, 2,3-epithiopropyl methacrylate (ETMA), was synthesized according to a previous procedure.26 The copolymer ligand of p(NIPAM-co-ETMA) was prepared by conventional free-radical copolymerization of NIPAM and ETMA monomers as follows: the polymerization was conducted in THF (15 mL), containing ETMA (0.2 g, 1.26 mmol), AIBN (0.016 g, 0.098 mmol), and NIPAM (2 g, 17.7 mmol) under inert atmosphere at 65 °C, using AIBN as initiator. After 12 h, the reaction solution was precipitated in petroleum ether, and the product was dried in vacuum at room temperature. 1H NMR (500 MHz, CDCl3, δ): 6.6 (b, 1H, CO –NH –CH(CH3)2),27 4.0 (c, 1H, CO–N –CH(CH3)2),28 3.7–3.8 (g, 2H, CH–CH2),29 3.61 (d, 1H, SCH–), 2.22–2.65 (i, 1H, SCH–CH2), 1.39–2.1 (a, 1H, CO–CH–CH2, e, 2H, –CH2), 1.16 (d, 3H, CO–NH–CH–CH3, CO–C–CH3).
Preparation of PNE-stabilized gold nanoparticles (Au NPs@PNE-a) with NaBH4 as reducing agent
All glassware used in the experiment was cleaned in a bath of freshly prepared aqua regia (HCl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :HNO3 = 3:1 by volume) and rinsed thoroughly in water prior to use. In a typical experiment, 1 mL HAuCl4 (0.005 M) was added to the PNE aqueous solution (0.03 mmol mL−1) with different molar concentrations under vigorous stirring (the samples from Au a-1 to Au a-7 represent PNE-capped Au NPs with different molar feed ratios of [HAuCl4]/[PNE] = 1:30, 1:20, 1:10, 1:5, 1:1, 1:0.5, and 1:0.1, based on the ratios of Au atoms to sulfur atoms in the copolymers). After 30 min, a certain concentration of NaBH4 solution was added, and the mixture was incubated at room temperature. The solution was then dialyzed in double-distilled water for a week and the final solution was stored at 4 °C before use.
:HNO3 = 3:1 by volume) and rinsed thoroughly in water prior to use. In a typical experiment, 1 mL HAuCl4 (0.005 M) was added to the PNE aqueous solution (0.03 mmol mL−1) with different molar concentrations under vigorous stirring (the samples from Au a-1 to Au a-7 represent PNE-capped Au NPs with different molar feed ratios of [HAuCl4]/[PNE] = 1:30, 1:20, 1:10, 1:5, 1:1, 1:0.5, and 1:0.1, based on the ratios of Au atoms to sulfur atoms in the copolymers). After 30 min, a certain concentration of NaBH4 solution was added, and the mixture was incubated at room temperature. The solution was then dialyzed in double-distilled water for a week and the final solution was stored at 4 °C before use.
Preparation of PNE-stabilized gold nanoparticles (Au NPs@PNE-b) by alkaline-induced reduction
Au NPs@PNE-b was synthesized using a modified procedure described in the preparation of Au NPs@PNE-a. In a typical synthesis, different pH values of hartshorn from 7 to 10, instead of NaBH4, was added to the mixture of HAuCl4 and PNE under vigorous stirring (the samples of Au NPs-b1, Au NPs-b2, and Au NPs-b3 represent PNE-capped Au NPs with molar feed ratios of [HAuCl4]/[PNE] = 1:30, 1:20, and 1:10, respectively). The solution was then dialyzed in double-distilled water for a week. The final obtained Au NPs were stored at 4 °C when not in use.
Catalytic experiments of Au NPs@PNE
The catalytic activity measurements of the Au NPs@PNE-a and Au NPs@PNE-b systems were based on the catalytic reduction of 4-nitrophenol to 4-aminophenol. In detail, 30 μL of 10 mM 4-nitrophenol, 2.77 mL of deionized water and 100 μL of the obtained Au NPs@PNE-a or Au NPs@PNE-b was added to a quartz cell as a substrate and incubated at a certain temperature in a water bath. Then, 100 μL of NaBH4 (0.00227 g mL−1) was added to the mixture. UV-vis spectroscopy was also conducted to record the system absorbance at 400 nm.
Measurements
Instrumental characterization. NMR spectra (1H-NMR and 13C-NMR) were obtained from an AVANCE Bruker spectrometer at basic frequencies of 500 MHz for 1H and 13C in CDCl3 solution. Thermogravimetric analysis (TGA) was conducted with a Perkin-Elmer TGA-2 thermogravimetric analyzer by heating the samples from ambient temperature to 800 °C with a heating rate of 10 °C min−1 in nitrogen. Differential scanning calorimetry (DSC) was performed on a TA DSC Q100 differential scanning calorimeter under a nitrogen atmosphere with a heating or cooling rate of 10 °C min−1. UV-vis absorption spectra were recorded on a SHIMADZU UV-2550 UV-visible spectrophotometer in the range 200–800 nm. The photoluminescence properties were measured on an FLSP920 Edinburgh Fluorescence Spectrometer. The molecular weight of the polymer was estimated at a flow rate of 1.0 mL min−1 at 25 °C by gel permeation chromatography (GPC) on a Waters instrument (Waters Corporation, USA), using CHCl3 as eluent, and the molecular weights were determined vs. polystyrene standards. Transmission electron microscopy (TEM) was carried out on a JEM-2100F electron microscope.
Results and discussion
Synthesis of p(NIPAM-co-ETMA) (PNE)
The novel copolymer ligand of p(NIPAM-co-ETMA) was prepared by conventional free-radical copolymerization of NIPAM and ETMA monomers, and its structure has been confirmed by FT-IR spectra and 1H-NMR. As shown in Fig. 1, the copolymer PNE and pure PNIPAM have similar FT-IR spectra due to the low feed ratio of ETMA monomers relative to NIPAM. The double peaks at 1653 and 1547 cm−1 are the typical characteristic vibrations of the amide I and amide II bands of PNIPAM, respectively.30 The stretching vibration of episulfide three-membered rings of ETMA units can be observed at 615 cm−1.31 In addition, a weak shoulder peak at 1734 cm−1, attributed to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration of ETMA units can be observed for PNE. The structure of the novel copolymer was further confirmed by NMR spectroscopy. In the 1H-NMR spectrum, different peak assignments are presented in Fig. 2. By comparing the integral area ratios of peaks h (the methine protons on the episulfide ring, –CH2CH(CH2)–S–) and c (the methine protons in PNIPAM block, CO–N–CH(CH3)2), the PNE structure has been confirmed and the molar ratio of NIPAM to ETMA units in PNE was calculated to be 5:1 by 1H NMR. The 13C-NMR spectrum of PNE is given in the ESI (see Fig. S1†). The 13C-NMR spectrum can provide qualitative information about the polymeric structure. The remaining NMR signals are matched with the structure. According to the DSC curve (Fig. S2†), the PNE exhibited a glass-transition temperature (Tg) between 121 and 129 °C. The thermal stability of PNE was studied by TGA and the TGA curve in Fig. S3† shows a single-step decomposition procedure, indicating that PNE is stable up to 240 °C. The obtained copolymers exhibited good solubility in different organic solvents (such as toluene, THF, chloroform, DMF, and DMSO) and aqueous solution. GPC measurements revealed that the number average molecular weight (Mn) of the copolymer is around 24000, and the copolymer has a relatively narrow molecular weight distribution with a polydispersity index (PDI) of 1.23.
O stretching vibration of ETMA units can be observed for PNE. The structure of the novel copolymer was further confirmed by NMR spectroscopy. In the 1H-NMR spectrum, different peak assignments are presented in Fig. 2. By comparing the integral area ratios of peaks h (the methine protons on the episulfide ring, –CH2CH(CH2)–S–) and c (the methine protons in PNIPAM block, CO–N–CH(CH3)2), the PNE structure has been confirmed and the molar ratio of NIPAM to ETMA units in PNE was calculated to be 5:1 by 1H NMR. The 13C-NMR spectrum of PNE is given in the ESI (see Fig. S1†). The 13C-NMR spectrum can provide qualitative information about the polymeric structure. The remaining NMR signals are matched with the structure. According to the DSC curve (Fig. S2†), the PNE exhibited a glass-transition temperature (Tg) between 121 and 129 °C. The thermal stability of PNE was studied by TGA and the TGA curve in Fig. S3† shows a single-step decomposition procedure, indicating that PNE is stable up to 240 °C. The obtained copolymers exhibited good solubility in different organic solvents (such as toluene, THF, chloroform, DMF, and DMSO) and aqueous solution. GPC measurements revealed that the number average molecular weight (Mn) of the copolymer is around 24000, and the copolymer has a relatively narrow molecular weight distribution with a polydispersity index (PDI) of 1.23.
 |
| | Fig. 1 FTIR spectra of p(NIPAM-co-ETMA) and pure PNIPAM. | |
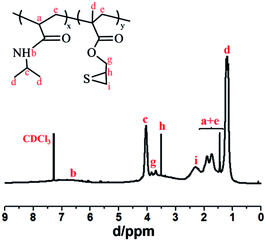 |
| | Fig. 2 1H-NMR spectrum of p(NIPAM-co-ETMA) in CDCl3. | |
Temperature-dependent optical transmissions at 600 nm for the copolymer in aqueous solution did not vary until about 27 °C (see Fig. 3). However, a sharp decrease in transmittance can be observed between 27 and 33 °C, which is associated with the “coil-to-globule” transition of the connecting PNIPAM chain segments.23 The phase transition turns the copolymer chain from hydrophilic to hydrophobic and breaks the interaction between the copolymer chains and the water molecules, because the PNE contains a predominant fraction of NIPAM units. The copolymer is no longer a homogeneous system under these conditions. So the PNE keeps its thermo-responsive properties, with a lower critical solution temperature (LCST) of around 29 °C in water, which is lower than that of pure PNIPAM. This result indicates that the incorporation of a hydrophobic co-monomer would reduce the amount of hydrophilic groups, which could result in a decrease in the hydrophilicity of the copolymer.32
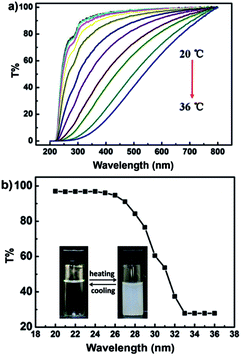 |
| | Fig. 3 Temperature dependence of transmission spectra (a) and the relative transmittance spectrum at 600 nm (b) of PNE in water. | |
We have tried two methods to produce small PNE copolymer-stabilized Au NPs, by direct in situ NaBH4 reduction of HAuCl4 in the presence of PNE, and an alkaline-induced reduction process. In this work, PNE, with episulfide ligands and thermo-responsive properties, was mainly used as a stabilizer for the controlled synthesis of Au NPs.
Synthesis of PNE-stabilized gold nanoparticles (Au NPs@PNE-a), with NaBH4 as reducing agent
When PNE was added to HAuCl4 solution, stirring for 30 min, it was observed that the yellow solution became gradually lighter, which may be caused by the interaction of Au3+ with sulfur atoms of PNE ligands in solution. It has been reported by Maeda that the ring-opening of the ETMA segment under acidic conditions can effectively chelate heavy metal ions.25 From Fig. 4, it can be observed that, half an hour after the interaction of Au3+ with PNE ligands, the absorption peak of PNE at 280 nm is no longer obvious and is replaced by a broad absorption curve. According to the change in solution color and the UV absorption spectrum, we speculate that the episulfide groups of ETMA units in the p(NIPAM-co-ETMA) copolymer may provide a site to form an Au3+-S-ETMA complex in PNE via episulfide-group ring-opening, and ring-opening caused by the strong acidic characteristics of HAuCl4.33
 |
| | Fig. 4 UV-vis absorption spectra of PNE aqueous solution before and after addition of Au3+. | |
The color changes of Au NPs@PNE-a systems with different Au/PNE ratios, after the addition of NaBH4, are shown in Fig. 5a. With an increasing ratio of [Au3+]/[PNE] from 1:30 to 1:0.1, the solution color gradually deepened, eventually becoming dark brown, which is related to the surface plasmon resonance of the formed gold NPs. This phenomenon indicated that, as the PNE content decreased, the size of the obtained gold NPs gradually increased, with a tendency to aggregate. Therefore, we believe that PNE is conducive to the stability of gold NPs, and a low ratio of [Au3+]/[PNE] results in smaller gold NPs. This conjecture is also supported by the UV-vis spectra in Fig. 5b. It can be seen that different ratios of Au/PNE-a systems correspond to different absorption wavelengths, suggesting that the gold NPs have different sizes.34,35 The obvious broad surface plasmon resonance (SPR) peak of the Au NPs can be observed at 520 nm when the Au/PNE ratios are higher than that of a3. The disappearance of the SPR absorption peak was found for Au NPs a1–a3, which may be attributed to the formation of smaller gold nanoclusters.36 This is because the small NPs no longer facilitate collective plasmon excitation due to the loss of metallic nature caused by the quantum confinement effect.37 The broad absorptions of a4 and a7 at about 520 nm are attributed to aggregates of these Au NPs, and this result corresponds with the colors of the solutions in Fig. 5a.
 |
| | Fig. 5 Digital images of Au NPs@PNE-a1 to a7 with different [Au3+]/[PNE] ratios in sunlight (a) and UV-vis absorption spectra of Au NPs@PNE-a1 to a7 with different [Au3+]/[PNE] ratios (b). | |
The particle size and properties were primarily controlled by varying the Au-to-ligand (Au:PNE) molar ratios when NaBH4 was used as reducing agent. Fig. 6 shows TEM images collected from different Au/PNE ratios for Au@PNE-a1, a3, a5, and a7 ([HAuCl4]/[PNE] = 1:30, 1:10, 1:1 and 1:0.1). It is noteworthy that the average size of gold NPs with a broad size distribution increases with decreasing PNE dosage. It is clearly seen from Fig. 6a that the maximum particle size of Au@PNE-a1 is close to 2.3 nm, while the minimum particle size for Au@PNE-a1 is only 0.93 nm. This result means that gold nanoclusters can be obtained from the reduction of Au3+-PNE by NaBH4. The maximum particle size of Au@PNE-a3 is 3.2 nm, while the average diameter of the other particles is about 1.5 nm. The average diameter of Au@PNE-a5 increases to about 3 nm as the ratio of Au3+/PNE increases. In addition, when [Au3+]/[PNE] is 1:0.1, the obtained gold NPs (Au@PNE-a7) are no longer spherical, and they display anisotropic growth characteristics, which may be because the low concentration of PNE ligands did not effectively stabilize gold nanoparticles, causing small particles with high surface ratios to aggregate to form a special morphology.
 |
| | Fig. 6 HRTEM (a and b) and TEM (c and d) images of Au NPs@PNE-a1 (a), a3 (b), a5 (c) and a7 (d). | |
Fig. 7 shows the fluorescence spectra of gold NPs (Au@PNE-a) with different Au3+/PNE ratios. It can be seen that when the [Au3+]/[PNE] ratio is less than 1:1 (Au@PNE-a1 to a5), the obtained gold NPs exhibit a distinct fluorescence emission peak at 450 nm and its intensity decreases with the content of PNE. When the ratio [Au3+]/[PNE] is more than 1:1, for Au@PNE-a6 and a7, the fluorescence is almost undetectable. Dickson et al. found that the relationship between the numbers of gold atoms and the fluorescence of gold nanoclusters, Au5 (UV-emission), Au8 (blue), Au13 (green), Au23 (red), and Au31 (near-infrared emission), could be easily explained by the dependence of fluorescence on the size of gold nanoclusters through the “jellium” model.38 Combining the results shown in Fig. 6, we speculated that the blue luminescence of Au@PNE-a systems could be attributed to the intrinsic emission of the core of gold nanoclusters, namely the results of electronic transitions within the gold nanoclusters.7 As Au NPs@PNE-a has a non-uniform particle size distribution, the existence of large particles may be the reason for the weak fluorescence intensity in Au NPs@PNE-a. Although the average particle size of Au NPs@PNE-a1 to a5 gradually increases, these Au NPs still have weak fluorescence due to the systems containing some ultra-small gold clusters.
 |
| | Fig. 7 Excitation (red line) and emission spectra of Au NPs@PNE-a1 to a7. | |
Synthesis of PNE-stabilized gold nanoparticles (Au NPs@PNE-b) by alkaline-induced reduction
The method of alkaline-induced reduction is a green way to prepare gold nanoparticles because it does not require adding extra reducing agent. We chose three [Au3+]/[PNE] ratios of 1:30, 1:20, and 1:10 to prepare gold nanoparticles (denoted as Au NPs@PNE-b1, b2 and b3) by adjusting the pH of the solution. Fig. 8 shows the fluorescence spectra of gold NPs obtained from solutions with different [Au3+]/[PNE] ratios by adjusting the pH between 7 and 10. It can be seen from Fig. 8A and C that when the [Au3+]/[PNE] ratio is 1:30 and 1:10, Au NPs@PNE-b1 and b3, with a maximum emission of approximately 450 nm, can be obtained by adjusting the pH values between 7 and 10, and the highest fluorescence intensity was observed at pH = 9. It is worth noting that in Fig. 8B, when the [Au3+]/[PNE] ratio is 1:20, the resultant Au NPs@PNE-b2 obtained from the solution with pH values from 7 to 8 has double fluorescence emission peaks at 460 and 530 nm. And when the pH values of the solutions are adjusted to 9 and 10, only a wide emission peak at 460 nm can be observed. We speculated that the Au3+-PNE-b2 complex was not completely reduced to Au0-PNE-b2 at pH 7 and 8. A part of Au3+-PNE-b2 was reduced to Au0-PNE-b2 by alkaline-induced reduction, and rapidly aggregated into gold nanoparticle cores, while some Au3+-PNE-b2 was not completely reduced to Au+-PNE-b2 and an Au+ layer was formed on the surface of the Au NPs. So we believed that the double-emission characteristics of Au NPs@PNE-b2 must originate from the intrinsic luminescence core as well as the electron transfer between the PNE ligand and unreduced Au+ on the surface of Au NPs. When the pH was increased to 9 and 10, the Au+-PNE-b2 complex was completely reduced to Au0-PNE-b2, and the intrinsic emission became a primary fluorescent peak. In order to verify that the surface of Au NPs@PNE-b2 still contained Au+, we added NaBH4 to the solution of Au NPs@PNE-b2 at pH = 7 and 8, and the fluorescence spectra of the resulting solutions are shown in Fig. 8D. It was found that the fluorescence emissions at 530 nm for both of the samples were quenched, while the fluorescence at 460 nm was retained. Therefore, the Au+ on the surface of Au@PNE-b2 could be reduced by the addition of NaBH4, which can block the electron transfer between Au+ and PNE, and thus the emission at 530 nm was quenched. Meanwhile, the addition of NaBH4 did not affect the core structure of Au NPs@PNE, thereby the intrinsic luminescence at 460 nm was maintained. This conjecture was also supported by the UV-vis absorption spectra shown in Fig. 9. It can be seen that the obvious SPR absorption at 530 nm was not observed for Au NPs@PNE-b2, obtained by alkaline-induced reduction at pH = 7–10. The reason may be that the gold particle size is less than 2 nm under alkaline-induced reduction, and this is not enough to support surface plasmon resonance. It is noteworthy that Fig. 9 shows an obvious absorption for Au NPs@PNE-b2 at 372 nm, which may be caused by charge transfer between the Au+ in the particle surface and the PNE ligands. However, this absorbance disappeared when the reducing agent, NaBH4, was added to the solution of Au NPs@PNE-b2 at pH = 7. This may be because the surface Au+ of Au NPs@PNE-b2 was reduced, blocking the charge transfer of Au+-PNE-b2. As discussed regarding Fig. 8, this result also indicates that the fluorescence of Au NPs@PNE-b2 originates from the synergistic effect of the intrinsic emission of Au NPs and the charge transfer emission of Au+-PNE-b2 on the surface of particles.
 |
| | Fig. 8 PL spectra of Au NPs@PNE-b1 (A), b2 (B), and b3 (C) prepared at different pH and PL spectra of Au NPs@PNE-b2 before (solid line) and after (dotted line) addition of NaBH4 (D) to Au NPs@PNE-b2 (pH = 7). | |
 |
| | Fig. 9 UV-vis absorption spectra of Au NPs@PNE-b2 at different pH [insert: UV-vis absorption spectra of Au NPs@PNE-b2 at pH = 7 before (black line) and after (green line) addition of NaBH4]. | |
The composition of Au NPs@PNE-b was further checked using XPS. Fig. 10 represents the XPS spectra of Au NPs@PNE-b1 and Au NPs@PNE-b2. For Au NPs@PNE-b1 (pH = 7), only Au(0) peaks are observed at binding energies of 87.2 eV (Au-4f5/2) and 83.6 eV (Au-4f7/2).39 Therefore, almost all the gold atoms on the surface of Au NPs@PNE-b1 (pH = 7) exist in the form of Au0. As shown in Fig. 11b, Au NPs@PNE-b2 (pH = 7) exhibits a binding energy (BE) at 84.3 eV for Au 4f7/2, which falls midway between Au(0) (BE = 83.7 eV) and Au(I) (BE = 85.1 eV). Therefore, this shows that Au(I) and Au(0) are present on the surface of Au NPs@PNE-b2.40 On the basis of the ratio of areas between the two 4f7/2 peaks, it can be concluded that approximately 37% of the Au is on the surface of the Au NPs@PNE-b2 as Au(I). This result is also in good agreement with our conjecture that both the Au NPs cores and the Au+ on the surface of Au@PNE-b2 are responsible for the double-channel emission from the Au hybrid NPs.
 |
| | Fig. 10 XPS spectra of (A) Au NPs@PNE-b1 (pH = 7) and (B) Au NPs@PNE-b2 (pH = 7). The original spectrum is in black, the fitted spectrum is in yellow, the Au+ 4f spectrum is in blue, and the Au0 4f spectrum is in green. | |
 |
| | Fig. 11 HRTEM images of Au NPs@PNE-b2 before and after addition of NaBH4. | |
Fig. 11a shows the TEM images of Au NPs@PNE-b2 (pH = 7). The average diameter of the gold NPs is 1.06 nm. This result is consistent with the speculation regarding Fig. 8, and the emission at 450 nm for Au NPs@PNE-b2 (pH = 7) should originate from the intrinsic luminescence of the cores of gold particles. Fig. 11b shows a TEM image of Au NPs@PNE-b2 (pH = 7) after adding NaBH4 to the solution of particles. It can be seen that, before and after NaBH4 was added, the size of Au NPs@PNE-b2 increased slightly, but there was no aggregation phenomenon. This may be caused by the reduction of the Au+ on the surface of Au@PNE-b2, and then the gold deposited on the surface of Au NPs@PNE-b2. The above discussions also indirectly indicated the existence of Au+ on the surface of Au NPs@PNE-b2.
Catalytic ability of thermo-responsive PNE-stabilized Au NPs for 4-NP reduction
The catalytic activity of Au NPs@PNE was studied through a well-known model reaction, which is the reduction of 4-nitrophenol (4-NP) to 4-aminophenol (4-AP) in the presence of NaBH4. Initially, the 4-NP solution has a bright yellow color, which turns yellow-green upon the addition of NaBH4. In addition, the absorption peak of 4-NP immediately undergoes a red shift from 317 to 400 nm, owing to the formation of 4-nitrophenolate ions in alkaline conditions by the action of NaBH4 (see Fig. S4†).41 As shown in Fig. S5,† without addition of the catalyst Au NPs@PNE, the absorption peak at 400 nm remains undiminished for a long time, demonstrating that the reducing agent, NaBH4, is unable to reduce the 4-nitrophenolate ion itself. However, with the use of Au NPS@PNE, the dark yellow color gradually became colorless with the progression of the reduction to 4-AP (see Fig. S6†). The kinetics of the reaction can be intermittently monitored using UV-vis spectroscopy. We selected Au@PNE-b2 (pH = 7), Au@PNE-b2 (pH = 9), Au@PNE-a3, and Au@PNE-a7 as catalysts to study their catalytic activity in the reduction of 4-NP. The reaction process was tracked by UV-vis measurement, as shown in Fig. 12. Specifically, as seen in Fig. 12, the decrease in the UV absorption intensity at 400 nm is determined by the reduction of 4-NP. In addition, the peak at 317 nm can be attributed to the production of 4-AP. The rate constant (k) of the reaction can be determined from the plot of ln(Ct/C0) vs. time.42,43 There exists a clear isosbestic point between the two absorption bands in the UV spectra, indicating that the two primary species are responsible for the conversion reaction. On the basis of the UV-vis spectra, therefore, pseudo-first-order reaction kinetics are applied to determine the rate constant k for the reaction.44 From Fig. 12A and D, the reduction reaction finished in 3.5 and 26 min with k = 0.632 and 0.044 min−1 for Au NPs@PNE-b2 (pH = 7) and Au NPs@PNE-a7 catalyzed reaction systems, respectively. From Fig. 14, we can determine the rate constant (k) of Au NPs@PNE-b2 (pH = 7) at 25 °C to be 0.632 min−1. From Fig. 12B and C, we can see that when Au NPs@PNE-b2 (pH = 9) and Au NPs@PNE-a3 were used as the catalysts, the catalytic reduction reaction was not complete at 30 min and 20 min, respectively, indicating that the catalytic efficiency of the two catalysts is very low.
 |
| | Fig. 12 Successive UV-vis absorption spectra of the NaBH4 reduction of 4-AP catalyzed by: (A) Au NPs@PNE-b2 (pH = 7), (B) Au NPs@PNE-b2 (pH = 9), (C) Au NPs@PNE-a3, and (D) Au NPs@PNE-a7. | |
Based on the experimental results above, Au NPs@PNE-b2 (pH = 7) and Au NPs@PNE-a7 showed good catalytic activity in the catalytic reduction of 4-NP. However, Au NPs@PNE-b2 (pH = 9) and Au NPs@PNE-a3 did not show a strong catalytic effect in the catalytic reduction of 4-NP. Through analysis of the morphology and structure of Au NPs@PNE-b, we speculate that Au+ on the surface of Au NPs@PNE-b2 plays an important role in the catalytic performance of Au NPs@PNE-b2 (pH = 7). In alkaline solution, 4-AP is converted to 4-nitrophenolate ions. Au+ can be adsorbed on the 4-nitrophenolate ions by electrostatic interaction, and then 4-nitrophenolate ions can form effective mutual interactions with the gold particle core to promote the reduction of 4-NP to 4-AP (see Scheme 3). It was found that for the Au NPs@PNE-b2 (pH = 7) with Au+ on its surface, the order of addition of the reducing agent affects the catalytic activity. Because, if the reducing agent, NaBH4, is added first in the 4-AP solution, the surface Au+ will be reduced to Au0 after the addition of Au NPs@PNE-b2 (pH = 7), and 4-nitrophenolate ions cannot be effectively adsorbed on the Au+ on the surface of Au NPs, which results in a loss of catalytic activity (see Fig. 13). This result confirms that Au+ on the surface of Au NPs plays an important role in the catalytic performance of Au NPs@PNE-b2 (pH = 7). It further demonstrates that there is the oxidation state of gold atoms on the surface of Au NPs@PNE-b2 (pH = 7). However, Au NPs@PNE-b2 (pH = 9) and Au NPs@PNE-a3 without Au+ on their surface did not effectively adsorb 4-nitrophenolate ions because the Au0 ground state electron configuration is 4f145d106s1 and the structure of the d orbitals is completely filled due to lower catalytic activity.45 Therefore, the catalytic activities of Au NPs@PNE-b2 (pH = 9) and Au NPs@PNE-a3 are lower than that of Au NPs@PNE-b2 (pH = 7). Regarding the catalytic activity of Au NPs@PNE-a3 and Au NPs@PNE-a7, it can be seen from the TEM images (Fig. 6) that Au NPs@PNE-a7 is no longer spherical, and it displays anisotropic growth characteristics. According to previous reports,46–48 anisotropic gold nanoparticles usually have better catalytic ability than spherical nanoparticles. In addition, the rate of electron transfer at the metal surface can be influenced by two aspects: (1) diffusion of 4-NP to the metal surfaces and (2) interfacial electron transfer and diffusion of 4-AP away from the surface.49 Thus, the diffusion of 4-NP should mainly determine the rate of the reduction. The adsorbing copolymer would affect the diffusion of 4-NP to the surface of metal nanoparticles. Thus, from this point of view, the high catalytic efficiency of Au NPs@PNE-a7 should be attributed to it having the least coating of PNE on the surface of Au NPs as compared with Au NPs@PNE-a3. Similar results have also been found in other research systems.49–51
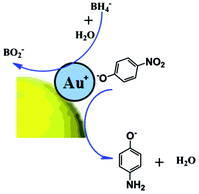 |
| | Scheme 3 Postulated reaction pathways of 4-nitrophenol reduction catalyzed by Au NPs@PNE-b2 (pH = 7). | |
 |
| | Fig. 13 Successive UV-vis absorption spectra of 4-AP after addition of NaBH4, followed by Au NPs@PNE-b2 (pH = 7). | |
The PNIPAM chains in the PNE copolymer can change their position with the shrinking and extension of the PNIPAM chains under thermal stimuli.52 So, the thermo-responsive catalytic behavior of Au NPs@PNE-b2 (pH = 7) was also investigated by UV-vis absorption spectroscopy. As shown in Fig. 14, the catalytic systems containing Au NPs@PNE-b2 (pH = 7) were subjected to an increasing temperature up to the LCST of PNIPAM in 80 s, and at about 180 s, the temperature was dropped to below the LCST of the PNIPAM. Fig. 14 shows the k values at different times. Clearly, the value of k increases with temperature in the range below the LCST, indicating that the rate of reduction can be enhanced by increasing the temperature. This tendency follows the typical dependence of the rate constant on temperature described by the Arrhenius equation, and is similar to that of general catalysts.53 As the temperature further increased, the rate constant k decreased and tended to 0 from 150 s to 180 s, which was the “frozen” state during the reaction time. When the temperature was reduced to below the LCST, the catalytic reduction of 4-AP began to “thaw”, and tended to be stable. As shown in Scheme 4, when the reaction temperature is raised to above the LCST, the opposite phase inversion occurred for the polymer ligand on the surface of Au@PNE-b2 (pH = 7), and the value of k decreased as the temperature increased until it reached a constant at about 33 °C. The abnormal decrease in the value of k is possibly due to the increase in temperature causing PNIPAM to become hydrophobic, and then the PNIPAM chains collapsed to form a hydrophobic barrier on the gold NPs at this temperature, which inhibited the access of the reactants to the gold NPs. Therefore, the adsorption of 4-nitrophenolate ions and the reducing agent on the Au+ surface is hindered. When the temperature was reduced below the LCST, the surface of the PNE-stabilized gold NPs recovered their stretched state, and the surface channels of the gold NPs were opened, which permitted contact of the reducing agent with 4-nitrophenolate ions, so that the reduction reaction continued. Thus, the thermo-responsive PNIPAM chains act as switches, which can be used to control the reaction by adjusting the temperature.
 |
| | Fig. 14 Plots of ln(Ct/C0) vs. time at different temperatures in the range below and above the LCST, when Au NPs@PNE-b2 (pH = 7) was used as catalyst. | |
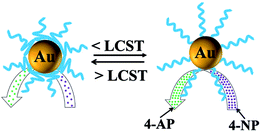 |
| | Scheme 4 Responsive catalysis of thermo-responsive copolymer-stabilized gold NPs, where the reduction of 4-NP by NaBH4 was chosen as a model reaction. | |
Conclusions
In summary, the well-defined novel thermo-responsive copolymer, p(NIPAM-co-ETMA) (PNE), was prepared by free-radical copolymerization. The copolymer-stabilized gold NPs could be further obtained by an Au3+-induced episulfide opening reaction and in situ reduction of HAuCl4 using NaBH4 and alkali as reducing agents in the presence of PNE. We found that Au NPs@PNE-a systems (NaBH4 as reducing agent) exhibited blue luminescence originating from the intrinsic emission of the gold particle core. The double-channel-emitting gold nanoclusters can be obtained by an alkaline-induced reduction route for the first time. The multiple colors could be attributed to the intrinsic emission and charge transfer emission of Au+ on the surface of Au NPs@PNE-b2, since the alkaline-induced reduction is not sufficient to produce this. Au NPs@PNE-b2 and the anisotropic Au NPs@PNE-a7 had a certain catalytic activity for the reduction of 4-AP. In addition, we found that the Au+ on the surface of gold NPs could effectively promote the reduction of 4-AP, resulting in a higher catalytic activity. Because of the thermo-responsive behavior of PNE, the catalytic activity of Au NPs@PNE also exhibited a thermal switch effect. Thus, the thermo-responsive PNIPAM chains played a role in the control of the catalytic rate. Our study showed that the copolymer played a vital role in the process of preparing gold nanoparticles with high catalytic activity and good stability. Therefore, these novel copolymers containing episulfide ligands may find many potential applications in the control of catalytic activity, catalyst recycling, and as ratiometric fluorescent probes with single-component Au NPs.
Acknowledgements
We appreciate the financial support of the National Natural Science Foundation of China (21574017).
Notes and references
- F. Vatansever, W. C. M. A. de Melo, P. Avci, D. Vecchio, M. Sadasivam, A. Gupta, R. Chandran, M. Karimi, N. A. Parizotto and R. Yin, FEMS Microbiol. Rev., 2013, 37, 955–989 CrossRef CAS PubMed.
- M. Hu, J. Chen, Z. Y. Li, L. Au, G. V. Hartland, X. Li, M. Marquez and Y. Xia, Chem. Soc. Rev., 2006, 35, 1084–1094 RSC.
- O. V. Salata, J. Nanobiotechnol., 2004, 2, 3 CrossRef PubMed.
- H. Otsuka, Y. Nagasaki and K. Kataoka, Adv. Drug Delivery Rev., 2012, 64, 246–255 CrossRef.
- Y. Mei, Y. Lu, F. Polzer, M. Ballauff and M. Drechsler, Chem. Mater., 2007, 19, 1062–1069 CrossRef CAS.
- R. Jin, Nanoscale, 2010, 2, 343–362 RSC.
- J. Zheng, C. Zhou, M. Yu and J. Liu, Nanoscale, 2012, 4, 4073–4083 RSC.
- L. Shang and G. U. Nienhaus, Biophys. Rev., 2012, 4, 313–322 CrossRef CAS.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- X. Le Guével, B. Hötzer, G. Jung and M. Schneider, J. Mater. Chem., 2011, 21, 2974–2981 RSC.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- G. Li and R. Jin, J. Am. Chem. Soc., 2014, 136, 11347–11354 CrossRef CAS PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS PubMed.
- F. Goto, K. Ishihara, Y. Iwasaki, K. Katayama, R. Enomoto and S. i. Yusa, Polymer, 2011, 52, 2810–2818 CrossRef CAS.
- L. Shang, S. Dong and G. U. Nienhaus, Nano Today, 2011, 6, 401–418 CrossRef CAS.
- S. Berchmans, P. J. Thomas and C. Rao, J. Phys. Chem. B, 2002, 106, 4647–4651 CrossRef CAS.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed.
- A. Thomas, Chem. Commun., 2012, 48, 6845–6847 RSC.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- H. Hamamoto, Y. Suzuki, Y. Yamada, H. Tabata, H. Takahashi and S. Ikegami, Angew. Chem., Int. Ed., 2005, 117, 4612–4614 CrossRef.
- J. Zhang, M. Zhang, K. Tang, F. Verpoort and T. Sun, Small, 2014, 10, 32–46 CrossRef CAS PubMed.
- N. Yan, J. Zhang, Y. Yuan, G. T. Chen, P. J. Dyson, Z. C. Li and Y. Kou, Chem. Commun., 2010, 46, 1631–1633 RSC.
- H. Wei, S. X. Cheng, X. Z. Zhang and R. X. Zhuo, Prog. Polym. Sci., 2009, 34, 893–910 CrossRef CAS.
- Y. Wang, G. Wei, W. Zhang, X. Jiang, P. Zheng, L. Shi and A. Dong, J. Mol. Catal. A: Chem., 2007, 266, 233–238 CrossRef CAS.
- H. Egawa, Anal. Chim. Acta, 1984, 162, 339–346 CrossRef.
- T. Nonaka and K. Fujita, J. Membr. Sci., 1998, 144, 187–195 CrossRef CAS.
- L. Y. Li, W. D. He, W. T. Li, K. R. Zhang, T. T. Pan, Z. L. Ding and B. Y. Zhang, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 5018–5029 CrossRef CAS.
- Y. Zhao, X. Shi, H. Gao, L. Zhang, F. Zhu and Q. Wu, J. Mater. Chem., 2012, 22, 5737–5745 RSC.
- M. L. Tebaldi de Sordi, M. A. Ceschi, C. L. Petzhold and A. H. E. Müller, Macromol. Rapid Commun., 2007, 28, 63–71 CrossRef CAS.
- Y. Tang, L. Liu, J. Wu and J. Duan, J. Colloid Interface Sci., 2013, 397, 24–31 CrossRef CAS PubMed.
- C. Lü, Z. Cui, Y. Wang, B. Yang and J. Shen, J. Appl. Polym. Sci., 2003, 89, 2426–2430 CrossRef.
- L. Hou and P. Wu, Soft Matter, 2015, 11, 2771–2781 RSC.
- Z. Luo, V. Nachammai, B. Zhang, N. Yan, D. T. Leong, D. E. Jiang and J. Xie, J. Am. Chem. Soc., 2014, 136, 10577–10580 CrossRef CAS PubMed.
- A. V. Gaikwad, P. Verschuren, E. Eiser and G. Rothenberg, J. Phys. Chem. B, 2006, 110, 17437–17443 CrossRef CAS PubMed.
- V. Amendola and M. Meneghetti, J. Phys. Chem. C, 2009, 113, 4277–4285 CAS.
- M. Nawaz, I. Ud-Din, G. Price and M. K. Baloch, J. Polym. Res., 2014, 21, 1–7 CAS.
- Z. Wu, J. Suhan and R. Jin, J. Mater. Chem., 2009, 19, 622–626 RSC.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- T. F. Jaramillo, S. H. Baeck, B. R. Cuenya and E. W. McFarland, J. Am. Chem. Soc., 2003, 125, 7148–7149 CrossRef CAS PubMed.
- B. Liu, Y. Wang, M. Deng, J. Lu, C. Tong and C. Lü, RSC Adv., 2014, 4, 57245–57249 RSC.
- Y. C. Chang and D. H. Chen, J. Hazard. Mater., 2009, 165, 664–669 CrossRef CAS PubMed.
- A. Murugadoss and A. Chattopadhyay, Nanotechnology, 2008, 19, 015603 CrossRef CAS PubMed.
- X. Chen, D. Zhao, Y. An, L. Shi, W. Hou and L. Chen, J. Nanopart. Res., 2010, 12, 1877–1887 CrossRef CAS.
- E. Seo, J. Kim, Y. Hong, Y. S. Kim, D. Lee and B. S. Kim, J. Phys. Chem. C, 2013, 117, 11686–11693 CAS.
- J. C. Fierro Gonzalez, J. Guzman and B. C. Gates, Top. Catal., 2007, 44, 103–114 CrossRef CAS.
- J. Zeng, Q. Zhang, J. Chen and Y. Xia, Nano Lett., 2009, 10, 30–35 CrossRef PubMed.
- H. G. Liao, Y. X. Jiang, Z. Y. Zhou, S. P. Chen and S. G. Sun, Angew. Chem., Int. Ed., 2008, 120, 9240–9243 CrossRef.
- Q. Cui, B. Xia, S. Mitzscherling, A. Masic, L. Li, M. Bargheer and H. Möhwald, Colloids Surf., A, 2015, 465, 20–25 CrossRef CAS.
- Y. Liu, L. Liu, M. Yuan and R. Guo, Colloids Surf., A, 2013, 417, 18–25 CrossRef CAS.
- Y. Bao, G. Shen, H. Liu and Y. Li, Polymer, 2013, 54, 652–660 CrossRef CAS.
- S. Panigrahi, S. Basu, S. Praharaj, S. Pande, S. Jana, A. Pal, S. K. Ghosh and T. Pal, J. Phys. Chem. C, 2007, 111, 4596–4605 CAS.
- T. Yin, X. Liu, J. Wang, Y. An, Z. Zhang and L. Shi, RSC Adv., 2015, 5, 47458–47465 RSC.
- Y. Wang, G. Wei, F. Wen, X. Zhang, W. Zhang and L. Shi, J. Mol. Catal. A: Chem., 2008, 280, 1–6 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17690a |
| ‡ These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 b,
Jianhua Lüa and
Changli Lü*a
b,
Jianhua Lüa and
Changli Lü*a

















