
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mesoporous carbon soft-templated from lignin nanofiber networks: microphase separation boosts supercapacitance in conductive electrodes†
Mariko
Ago
*a,
Maryam
Borghei
a,
Johannes S.
Haataja
b and
Orlando J.
Rojas
*ab
aBio-Based Colloids and Materials and Centre of Excellence on “Molecular Engineering of Biosynthetic Hybrid Materials Research” (HYBER), Department of Forest Products Technology, Aalto, University, FIN-00076, Espoo, Finland. E-mail: mariko.ago@aalto.fi; orlando.rojas@aalto.fi
bMolecular Materials, Department of Applied Physics, Aalto University, FIN-00076, Espoo, Finland
First published on 30th August 2016
Abstract
Flexible electrodes with supercapacitance were developed from highly mesoporous carbon fibers synthesized from lignin. Polyvinyl alcohol (PVA) facilitated the electrospinning of aqueous solutions of lignin and was used as a sacrificial polymer. Most importantly, PVA produced phase-separated domains for extreme surface area (>2000 m2 g−1) and mesoporous volume (0.7 cm3 g−1). An optimized sequential thermal treatment that initially included stabilization at 250 °C, allowed the formation of flexible, freestanding carbon networks upon PVA evolution to the gas phase and carbonization of the as-spun lignin-based fibers. Their main morphological and chemical characteristics were assessed by field emission scanning microscopy, transmission electron tomography reconstructions and Raman spectroscopy. The carbon fiber networks were used directly as electrodes with electrochemical double layer capacitance as determined by cyclic voltammetry and galvanostatic charge/discharge methods. Excellent electrochemical performance was demonstrated from the measured high rate capability and long-term cycling stability. The determined specific capacitance (∼205 F g−1 in 0.5 M Na2SO4 electrolyte) is one of the highest recorded for electrodes obtained from biopolymer precursors. Moreover, the electrical conductivity of the carbon fiber network (386 S m−1) was significantly higher, by two-orders of magnitude, than that obtained from the precursor (non-fibrous, powder) sample (2.47 S m−1). The remarkable performance of the synthesized electrodes is ascribed to the robust network morphology and mesoporosity obtained by soft-templating from the phase-separated sacrificial polymer. This is a demonstration of lignin valorization for novel application in advanced materials.
Introduction
Electric double layer capacitors (EDLC), the most common and current devices for energy storage, are based on the electrical adsorption of ions at the electrode/electrolyte interface. EDLCs have a number of desirable characteristics, such as high power and energy density as well as rapid charge/discharge capability. Supercapacitors have the potential to compete with or replace batteries for energy conversion and storage, this is most relevant to transportation, including hybrid electric vehicles and metro-train transportation.1,2 Carbon materials, including activated carbon,3–7 and those derived from aerogels,8 nanotubes,9,10 templated porous systems,11–13 and nanonofibers,14 have been considered for related purposes. This is because of advantages that include low cost, easy processing, non-toxicity, high specific surface area, good electric conductivity, high chemical stability, and a wide range of operational temperatures.15 However, there are still technical challenges that need to be overcome for developing high electrical capacitance.Carbons for double-layer type supercapacitors should ideally have three main properties, namely, (1) high specific surface areas, of the order of 1000 m2 g−1, (2) good intra- and inter-particle conductivity in porous matrices, and (3) good electrolyte accessibility to the intrapores.16 Several methods have been developed to obtain large specific surface areas as well as suitable porosity in carbon materials, including activation in alkaline or acidic media,17–19 steam20,21 or carbon dioxide treatments,22 hard-templating via filling of inorganic particles within an organic precursor, organic–organic assembly via soft-templating,23,24 among others. Particularly, soft-templating methods are suitable for developing mesoporosity in carbon materials towards conductive electrodes. This is mainly because of the small amounts of impurities left behind after carbonization.
Prior studies have reported successfully on the fabrication of ordered mesoporous carbons by using soft-templating synthesis with thermosetting network polymers. This has included phenolic-type resins as a carbon source and block copolymers; resorcinol–formaldehyde and diblock copolymers (PS-b-P4VP);25 resorcinol–formaldehyde and Pluronic surfactants26,27 and PAN-b-PMMA.28 In these studies, the pore size developed from the precursor structure was well controlled, at the molecular scale.
However, some drawbacks exist in these systems, such as the complex procedure used for polymer synthesis, time-consuming protocols and relatively high cost. Furthermore, additional limitations include the choice of both carbon and porogens precursor materials, to avoid the collapse of the porous structure during high-temperature treatment. In fact, only few materials meet associated requirements.23 In this context, lignin has attracted attention as a precursor of carbon materials owing to its high carbon content (more than 60% in phenyl propane groups), as well as its thermal stability, and favorable stiffness.29,30 Thus, we propose lignin as a widely available (exceeding 300 billion tons31 as a byproduct of the pulp and paper industries and biorefineries),32 renewable and inexpensive aromatic polymer for the generation of carbon electrodes and as an attractive option to replace synthetic thermosetting polymers as source of mesoporous carbon. Soft-templating, on the other hand, can be achieved by addition of a sacrificial polymer, polyvinyl alcohol (PVA).
The few, recent studies related to supercapacitors by using activated carbon from lignin are listed in Table 1. A capacitance as high as 268 F g−1 (ref. 33) was recorded in the presence of a conductive additive in the working electrodes.
| Carbon source (reference) | Electrode and composition | Conductive additive | Electrolyte | Specific capacitance, F g−1 |
|---|---|---|---|---|
| a PVDF = polyvinylidene fluoride; PTFE = polytetrafluoroethylene; NEt4BF4 = tetramethyl ammonium tetrafluoroborate; CB = carbon black. “Carbon” in the “Electrode” column refers to the material obtained from lignin. | ||||
| Kraft lignin13 | Carbon![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :comercial supercarbon:PVDF, 75:25:10 :comercial supercarbon:PVDF, 75:25:10 |
Commercial high-surface-area supercarbon | 6 M KOH | 102.3 |
| Black liquor33 | Carbon:carbon black:PTFE, 89:5:6 |
Commercial functionalized CB | 6 M KOH/LiOH (50/1, w/w) | 267.8 |
| Alkali lignin34 | Carbon fibrous mat | None | 6 M KOH | 64 |
| Black liquor35 | Carbon:PTFE, 95:5 |
None | 1.5 M NEt4BF4 | 87 |
| Kraft lignin, alkali extracted lignin36 | Carbon:conductive carbon black:PVDF, 95:5:5 |
Commercial conductive carbon black | 1 M H2SO4 | 114.4 |
| Alkali lignin (present work) | Carbon fibrous mat | None | 0.5 M Na2SO4 | 205 |
In this study we developed a highly mesoporous nano- and micro-carbon fiber network as conductive electrode for supercapacitance by using lignin as a carbon precursor. The distinct advantage of the proposed approach is the synthesis of a mesoporous carbon network that can be utilized directly as an electrode with no need of additives and even in the absence of binders.
Recently, we demonstrated that the sub-micron fibers can be synthesized easily by electrospinning of aqueous solution of lignin. We also concluded that the physical properties (fiber diameter, morphology, etc.) can be tuned by the addition of polyvinyl alcohol (PVA) as a minor component.37,38 Moreover, our previous studies indicated microphase separation between lignin and PVA, which spontaneously segregated during the electrospinning process.38 For the system containing 75 wt% lignin (PVA, 25 wt%), discontinuous lignin/PVA phases formed within a lignin-rich, external phase. Based on these results, we hypothesize that highly mesoporous carbon fiber networks are feasible upon carbonization and by templating the two-phase morphology, from microphase separation of lignin and PVA. Here, the main lignin domain would act as carbon scaffold, while the mesopore domain would result from the PVA phases acting as a soft template. Fig. 1 illustrates the proposed component distribution in lignin/PVA electrospun fibers.
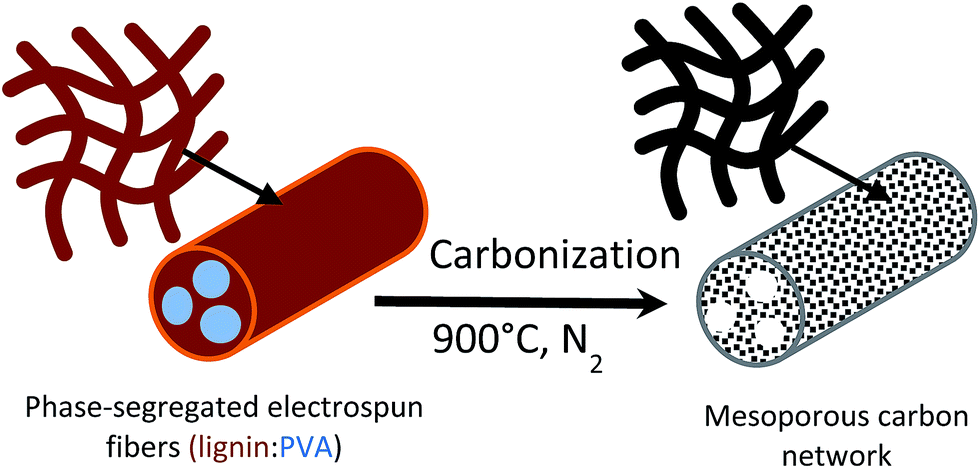 | ||
| Fig. 1 Schematic two-phase morphology of electrospun fiber (left) and mesoporous carbon (right). | ||
Free-standing fibrous carbon mats can be useful as performance conductive electrode in supercapacitors. A continuous fibrous morphology would also enable mechanical robustness, flexibility and stability. Furthermore, flexible electrodes, which may operate flawlessly even upon twisting, are desirable in the design of light and flexible devices, which are demanded in applications such as flexible circuits,39 displays,40 solar cells,41,42 and pressure sensors.43,44
The electrospinning technique has been widely adopted for fabricating fibrous mats from various polymer solutions.45–47 Such process balances electrostatic repulsive forces in the polymer solution and an externally-applied electric filed. Additionally, it can be scaled up to ensure low production costs. Here, we propose that high surface area electrospun mats obtained from lignin/PVA solutions, which are flexible and mechanically robust, are suitable, directly after carbonization, as freestanding electrodes. Therefore, we produced electrospun mats from lignin/PVA (75:25) as carbon precursor to fabricate highly mesoporous carbon networks using soft-templating. The morphology and porous structure of the resultant carbon network were investigated. In addition, electrochemical measurements for supercapacitance was performed to afford new storage devices from such mesoporous system. Overall, this contribution attempts to demonstrate the use of a renewable biopolymer to synthesize fibrous networks for free-standing and flexible carbon mats that can be used as supercapacitor electrodes.
Results and discussion
Aqueous solutions of lignin/PVA, at given concentrations, were found to be suitable in the production of defect-free electrospun fibers.37 In this study, a lignin/PVA mass ratio of 3:1 (w/w) and 17.4% (lignin + PVA) polymer concentration was chosen as precursor system for electrospinning. Mat 1, Mat 2, and Mat 3 are used herein to indicate the carbon fiber networks obtained by the respective carbonization protocol, as indicated in Table 2. The as-spun fibrous mats were subjected to carbonization at 900 °C for 2 h under nitrogen flow followed by different combinations of isothermal stabilization and temperature gradient. When the as-prepared electrospun mat was subjected to isothermal treatment at 250 °C, which is above Tg of lignin,48 the yield of the final carbon product was improved, up to 13%; in addition, the fiber morphology remained unchanged upon carbonization at 900 °C for 2 h (Mat 3). Without the stabilization step, the yield proved to be too low (Mat 0, ∼0% yield). The condensation reactions between phenolic units, which introduced the main lignin domain endowed fiber thermal stability.49,50 Without the stabilization step, the effect of decomposition, chain scission of lignin as well as PVA resulted in network disintegration (for a ∼0% yield upon carbonization at 900 °C). After the isothermal treatment, an equal amount of potassium hydroxide was used as a chemical activator to increase the specific surface area.17,51 The chemical activation was achieved during carbonization (up to 900 °C) under nitrogen flow. The heating rate used in the carbonization influenced the yield of the final product. A high heating rate, e.g., 20 °C min−1, was not suitable to produce a carbonaceous material (Mat 4, ca. ∼0% yield). However, a moderate heating rate (4–10 °C min) afforded yields between 10 and 13% (Mat 1, 2, and 3); it was also effective in retaining the nanostructure as well as fibrous network morphology. After many lignin carbonization tests, it was determined that freestanding fibrous carbon mats with preserved network structure can be achieved by thermal treatment (250 °C) for stabilization followed by heating to 900 °C (using a temperature gradient of 4 or 10 °C min−1). The three conditions for thermal treatment applied to the as-spun fiber mat afforded the highest efficiency in terms of yield and processing time. Also, compared to other conditions, they produced films with better flexibility and mechanical robustness.
| Sample | Isothermal treatment 250 °C, h | Heating rate, °C min | Yield, % | Morphology |
|---|---|---|---|---|
| Mat 0 | 0 | 5 | 0 | N/A |
| Mat 1 | 1 | 4 | 11 | Flexible mat |
| Mat 2 | 2 | 4 | 10 | Flexible mat |
| Mat 3 | 2 | 10 | 13 | Flexible mat |
| Mat 4 | 2 | 20 | 0 | N/A |
| Powder | 2 | 10 | 26 | Powder |
| Solid film | 2 | 10 | 45 | Brittle film |
The morphology of the as-spun fibers, before and after carbonization, is shown in Fig. 2. Fibers with a radius of 148 ± 8 nm were observed as a randomly oriented network. Fig. 2c and d show the morphology for Mat 3 after carbonization followed by isothermal treatment and chemical activation. Some fibers adopted a higher curvature and fused into the network structure possibly due to the melting of PVA distributed in the fibers during the isothermal treatment (250 °C). The diameter of the fibers remained unchanged after carbonization, as concluded from repeated experiments under the same conditions. A micrograph obtained with high magnification revealed numerous small pores on the surface of the fibers, which could be accessible to the electrolyte. The carbon mats could be bent easily (Fig. 2e) and recovered the original flat shape after the stress was release, indicating that the mats were mechanically strong and the fibers formed an entangled and interconnected network. As a reference, lignin powder was subjected to carbonization by using the same conditions as the ones used for Mat 3 (herein indicated as “Powder” sample). A lignin/PVA solid film prepared by solvent casting was also prepared for comparison (“Solid film”). Note that the lignin/PVA solid film is expected to be closer to thermodynamic equilibrium because of a “slow” consolidation upon solvent evaporation.38 In this slow process, the interfacial tension between lignin and PVA drives phase segregation to yield PVA domain sizes as large as 500 nm.38 The effect of the characteristic time of treatment on the porous structure of the derived carbon material (from electrospun fiber mats or solid films) is discussed later. Both the powder sample (powder) and the solid film (Solid film) presented a higher yield (26% and 45%, respectively) compared to the fibrous mats (Table 2).
 | ||
| Fig. 2 SEM images of electrospun lignin-based fiber mats before (a and b) and after carbonization at 900 °C, for 1 h at a rate of 10 °C min (c and d). Included also is a photograph of a flexible carbon mat (3 × 3 cm2) after carbonization (e). | ||
Nitrogen adsorption measurements (BET) were carried out in order to examine the specific surface area, pore volume, and pore size distribution of the carbon fibrous mats, powder and solid films (Table 3). The NLDFT model was used to analyze the BET isotherm.52 The pore size distributions for the powder and the mat samples are shown in Fig. 3. For the fibrous mats, highly specific surface areas were recorded (1689, 1387, and 2005 m2 g−1 for Mat 1, 2 and 3, respectively). Also, a high pore volume was determined (0.60, 0.71, and 0.70 cm3 g−1) with a dominant mesoporosity (58, 77, and 66%, respectively). The isotherms for the mat samples and the solid film presented a typical type-IV isotherm with a hysteresis loop in the P/P0 = 0.45–0.6 relative pressure region, which is attributed to mesoporous structure (Fig. S1a†). The isotherm displayed a rapid adsorption rise in the low-pressure region, indicating the presence of micropores. In contrast, the powder sample exhibited a BET specific surface area of 1236 m2 g−1 with total pore volume of 0.25 cm3 g−1 along with a half of mesoporous volume (51% mesopore). Besides, the isotherm of the powder sample showed type-I isotherm. These results suggest that the powder sample possessed a highly microspore structure (Fig. S1b†). The fibrous mats and the powder sample exhibited two prominent peaks in the pore size distribution, in the micropore (<2 nm) and mesopore (2–50 nm) regions. An interesting observation is that compared to the powder sample, the fibrous carbon mats presented a broader distribution in the mesopore region, centered around 2.5 nm (Mat 1 and Mat 3) or 2.9 nm (Mat 2). The BET surface area and pore size distribution confirmed the presence of highly mesoporous structures with high specific surface area. The pore size distribution can be explained in terms of microphase separation of the precursor electrospun fibers. In the 75:25 lignin/PVA system, the two-phase morphology arises from discontinuous PVA domains separated from the lignin, main phase.38 As the electrospun fibers were subjected to carbonization, the lignin scaffold was carbonized while the PVA phase evolved into gas, leaving behind a porous network. Further insights into the effect of the PVA domain size on the mesopore distribution can be gained from the solid films prepared by the evaporation-casting method. In this process, the characteristic time for densification is much longer than in electrospinning, which leads to conditions that are closer to equilibrium. In turn, this induces large phase-separated domains, with sizes of the order of 500 nm. BET surface and pore volume determinations for the solid films revealed significantly reduced values compared to the mat samples. Moreover, the isotherm exhibited type-IV profile characteristic of mesoporous structures (Fig. S1c†).
| Sample | V total, cm3 g−1 | V micro, cm3 g−1 | V meso, cm3 g−1 | Mesopore ratio, % | BET SSA, m2 g−1 |
|---|---|---|---|---|---|
| Mat 1 | 0.602 | 0.256 | 0.346 | 58 | 1689 |
| Mat 2 | 0.714 | 0.163 | 0.551 | 77 | 1387 |
| Mat 3 | 0.703 | 0.242 | 0.461 | 66 | 2005 |
| Powder | 0.252 | 0.121 | 0.131 | 51 | 1236 |
| Solid film | 0.031 | 0.068 | 0.024 | 77 | 217 |
 | ||
| Fig. 3 Pore size distribution for carbon mats (Mat 1, 2, and 3) and the powder sample. | ||
The results suggest a lower pore volume for the films compared to the electrospun fibers, owing to the larger PVA domains in the former structure. The PVA domains were effective in introducing the mesoporous structure in the solid film, depending on the domain size. The mesoporous structure of the carbon fibrous mat is evident in TEM micrographs and transmission electron tomography reconstructions (Fig. 4), which indicate the presence of numerous randomly distributed mesopores (the lighter areas in Fig. 4b) with diameter of ∼5 nm. They can be ascribed to the PVA domains that soft-templated as pores that, in turn, would allow electrolyte access from the outer surfaces (Fig. 4c). The results of SEM and TEM analyses are consistent with the results of nitrogen adsorption. It was later noticed that the capacitance strongly depended on the specific surface area as well as the pore size distribution of the electrode materials. Since higher accessibility of the electrolyte can contribute on higher charge accumulation at the electrode-electrolyte interface.53
 | ||
| Fig. 4 TEM micrograph of carbon fibrous (a); cross section slice from 3D (ET) reconstruction (b); isosurface model from segmented 3D reconstruction of Mat 3 (c). Scale bar is 100 nm in (a) and 40 nm in (b and c). Black dots are fiducial gold markers used for image alignment purposes in the ET reconstruction. | ||
Raman spectroscopy was applied to estimate the degree of graphitization of the carbonized samples (carbon fibrous mat as well as powder sample) (see Fig. S2†). Two Raman bands in the region between 1100 and 1800 cm−1 were distinctively observed for both the mat and powder samples. A strong band at 1590 cm−1, the G-band, and another band at 1350 cm−1, the D-band, were apparent. A similar peak position of the D and G peaks for the different samples was noted. The presence of graphitic bonding due to the periodic sp2 valance in crystalline carbonaceous material would strengthen the intensity of the G-band, while the D-band would reflect the presence of disorder and defective carbonaceous constituents. Therefore, the relative D-to-G band intensity ratio, ID/IG, can be related to the perfection of the graphitic layered structure, reflecting the graphitization degree of the material.54 The ID/IG ratios were similar for the samples tested, around 1.25–1.26, which indicates that the carbonization condition and the morphology of the samples (fibrous mat or powder) did not influence the graphitization.
A high electrical conductivity is of great significance in the development of electrodes for supercapacitors. The electrospun carbon mats derived from the lignin/PVA bi-component fibers were not completely graphitized; however, the electrical conductivity of the mat samples (386 S m−1) was significantly larger, by two orders of magnitude, than the one measured for the powder sample (2.47 S m−1). This can be explained by the morphology of the fiber network that provides a continuous pathway for effective electron transport and for a reduced electrochemical resistance in the electrode. In contrast, the powder samples contain micron/nano-sized carbon grains that may not have enough entanglement to yield a proper electron conductivity.
Electrochemical measurements
The supercapacitance of the carbon fibrous mats and powder samples was studied by a three-electrode cell configuration. Cyclic voltammograms (CV) were obtained at cell potential window from −0.2 to 0.8 V (vs. Ag/AgCl) at scan rates (ν) from 5 to 100 mV s−1 in 0.5 M Na2SO4 electrolyte. The as-prepared carbon mats were used directly as the working electrode, while in the case of the power sample a modified glassy carbon electrode was produced with a drop of carbon ink.Fig. 5 compares the CV curves from Mat 3 (a) and powder (b) at scan rates of 5 to 50 mV s−1. It can be seen that all of the CV profiles exhibited a nearly symmetrical rectangular shape at low scan rates. This corresponds to an ideal reversible capacitive behavior with good performance characteristics, fast ion diffusion and electron transfer rates for EDLC. Nevertheless, a distortion of the CV curves was observed at high scan rates, likely due to limitations in electrolyte transport and diffusion. Although this phenomenon can be seen quite often in carbon-based electrode materials, further studies are needed to overcome this effect. It is noticeable that the current density for Mat 3 was higher than that of the powder sample.
 | ||
| Fig. 5 Cyclic voltmmograms of Mat 3 (a) and powder (b) at various scan rates of 5, 10, 20, and 50 mV s−1. Galvanostatic charge–discharge curves of Mat 3 (c) and powder (d) at various current densities of, 0.3, 0.5, 1, and 2 A g−1 in 0.5 M Na2SO4. | ||
The capacitance values at a scan rate of 5 mV s−1 was calculated; under this condition a prominent rectangular shape in the CV plots, with maximum plateau flatness for both mat and powder samples, was observed. As expected, Mat 3 produced a higher specific capacitance, 205 F g−1, than that of the powder, 93 F g−1. The other samples, Mat 1 and Mat 2, exhibited capacitance values of 204 F g−1 and 155 F g−1, respectively. The remarkably high supercapacitance of the mat samples can be attributed to the high surface area and well-developed mesoporous structure, with open pores accessible to ion/electrolyte transport. Thus, the freestanding carbon network structure in the fibrous mats provides effectively more active sites due to high specific surface area and mesoporosity, as well as highly electrical conductivity. The featured high capacitance and the flexibility of the free-standing fibrous carbon mats offer promising advantages for use in electrochemical supercapacitor devices. At the same time, they involve facile and fast synthesis. As comparison, we further studied the Solid film that was prepared by solvent casting and noted a quite low capacitance (6.4 F g−1). Fig. 5c and d show typical charge–discharge profiles of Mat 3 and powder samples in 0.5 M Na2SO4 solution at current densities from 0.3 A g−1 to 2 A g−1. The charge–discharge curves were similar in shape between 0 and 0.6 V, indicating that the double layer capacitance can form reversibility in a wide range of current densities, for both mat and powder samples. Mat 3 electrode, with a large BET specific surface area, exhibited a longer discharging interval, which suggests a higher electrical capacity. The results were in accordance with the CV profiles. However, voltage drops at the turning point of charge–discharge can be seen, especially in the measurement with higher current density, i.e., 2 A g−1, for both mat and powder samples. This indicates energy losses due to the internal resistance by diffusion-limited mobility of the electrolyte ions in the electrode pores.
The cycling stability of the Mat 3 and powder samples was also examined by CV measurements over 1500 cycles, at a scan rate of 10 mV s−1, and the supercapacitance retention was monitored as a function of cycling number (Fig. 6). Mat 3 kept ∼83% of the initial supercapacitance after 1500 cycles, indicating good electrochemical stability as a supercapacitor electrode material. The fibrous carbon network in mat sample can work as a support matrix, which provide structural robustness and stability. For the powder sample, a steep decline in supercapacitance was observed, which may be due to the disintegration of active material and mechanical detachment from the glassy carbon electrode.
 | ||
| Fig. 6 Cycling stability of Mat 3 and powder electrodes (scan rate at 10 mV s−1). | ||
Experimental
Materials
Lignin was purchased from Sigma-Aldrich (St. Louis, MO) with reported molecular weight of 10 kDa (softwood alkali lignin, low sulfur, CAS Number: 8068-05-1). Poly(vinyl alcohol) (PVA), was acquired from the same supplier under trade name Mowiol® 20-98 (CAS Number: 9002-89-5), with reported molecular weight of 125 kDa and degree of hydrolysis of 98% (2% acetyl groups content). Both polymers were used without further purification to prepare aqueous solutions for electrospinning. Potassium hydroxide (Aldrich, Finland) and soap (Neutradish, Kiiltoclean, Finland) were used as received.Lignin/PVA blend electrospun fibers
Lignin/PVA solutions were prepared according to the method described in our previous report.37 Briefly, aqueous PVA solutions (15 wt% concentration) were prepared and lignin was added to obtain solutions with lignin/PVA dry mass ratio of 75:25. Distilled water was added to the lignin/PVA solution to obtain a viscosity suitable for electrospinning. The solution was kept under vigorous mechanical agitation at 60 °C for 1 h, followed by cooling to room temperature under stirring for 4 h. Solutions stored for less than one-week were used to produce the electrospun fibers. For electrospinning, the lignin/PVA solution was loaded into a 10 ml plastic, disposable syringe with a 22-gauge needle. The needle was connected to the positive terminal of a voltage generator designed to produce a voltage up to 30 kV DC (Spellman SL30, USA). A thin aluminum foil covering a 15 cm diameter aluminum plate was used as a collector. The plate was connected to the negative electrode of the power supply (ground) and set at a working distance of 20 cm. An operating voltage of 17 kV was used. A constant 8 or 10 μl min−1 flow rate was maintained during electrospinning using a computer-controlled syringe pump. Electrospinning was performed at room temperature and at 35–45% relative humidity. The collected electrospun mats were kept in a desiccator containing anhydrous silica gel.
Solid film preparation
Solid films were prepared by solution casting from lignin/PVA with a 75:25 dry mass ratio, same as used for the electrospinning. The aqueous solution was poured onto a clean Teflon plate and dried overnight at room temperature in a dust-free atmosphere. The resulting films were ca. 40 μm in thickness as detected by optical microscopy.
Carbonization
The carbonization procedure was optimized after a series of experiments. Firstly, stabilization of as-spun fiber was performed by heat treatment (heating rate of 4 °C min−1) at 250 °C for 1 or 2 h in a nitrogen atmosphere followed by a second heating treatment at 600 °C for 1 h. The stabilized fiber mats were impregnated with KOH in aqueous solution (fiber:KOH mass ratio of 1:1). The impregnated sample was dried in an oven at 60 °C for 2 h and then carbonized in a metal chamber inside a muffle furnace. Typically, the sample was heated to 900 °C for 2 h (heating rate of 4 °C min−1) under nitrogen gas flow (400 ml min−1). After the carbonization, the samples were cooled down to room temperature under nitrogen gas flow and washed with distilled water repeatedly until neutral pH followed by 1 M HCl rinsing and final washing with distilled water. As a last step, the samples were washed with ethanol and dried in an oven. Lignin powder was used as a control sample along with the respective solid films, both of which were subjected to the same procedure as indicated for the fibrous mats. These samples are referred to as “Powder” and “Solid film”.
Material characterization
The morphology of the electrospun nanofibers and powder samples were examined using a field emission scanning electron microscope (FE-SEM) (Zeiss SigmaVP, Germany) operating at 1.6 kV and a working distance 4 mm. A small piece of the nanofiber mat was fixed on a carbon tape and then sputtered with Pt. transmission electron microscopy (TEM) and electron tomography (ET) was carried out using JEOL JEM 3200FSC field emission microscopy operated at 300 kV in bright field mode with an omega-type zero-loss energy filter. For TEM measurements, the sample was dispersed in ethanol by sonication, and evaporated on a lacey carbon grid. ET tilt series acquirement was carried out with the SerialEM-software package by tilting the sample between ±70° with 2° increment steps. Image alignment was done with IMOD.55 Maximum entropy reconstruction scheme was carried out with MEM program56 on Linux cluster with regularization parameter value δ = 5 × 10−2. The images were binned twice to reduce noise and computation time. The specific surface area of the samples was analyzed by the Brunauer, Emmett, and Teller (BET) method while the micropore volume was estimated using the non-local density functional theory (NLDFT). Raman spectroscopy was used to detect the graphitic carbon structure and the Raman spectra of the carbonized samples were obtained by exposure to laser light (633 nm wavelength). The DC electrical conductivity of the carbonized samples (both carbon fiber mat and powder) was measured using a Jandel four-point probe system. The electrical conductivity σ was determined using the equation σ = L/ARS where Rs is the sheet resistance, A is the cross-sectional area, and L is the distance between the electrodes (6 × 10−3 m in our Jandel instrument).Electrochemical activity
The carbonized electrospun mats were used directly as a free-standing working electrode. For the powder sample, 40 mg ml−1 catalyst ink was prepared by dispersing the powder sample in ethanol containing ion exchange ionomer, FAA3, (Fuma-Tech) and deposited on a glassy carbon electrode followed by oven drying at 60 °C. The working electrodes were assembled into a three-electrode cell configuration with Ag/AgCl reference electrode and platinum plate or rod counter electrodes. Electrochemical evaluations were performed using an Autolab PGSTAT12 potentiostat (EcoChemie, The Netherlands) controlled by the GPE software. Cyclic voltammetry (CV) measurements were carried out at a potential window from −0.2 V to 0.8 V (0.5 M Na2SO4 electrolyte). Galvanostatic charge/discharge tests were also performed using various current densities (between 0.3 to 2 A g−1). The specific capacitance Cm was calculated from the discharging curves according to the following equation: , where I is the constant discharge current and t is the discharge time, m is the mass of active materials (g), and ΔV is the potential window of discharge. The cycling stability of the fibrous carbon mat and powder samples was carried out for over 1500 cycles.
, where I is the constant discharge current and t is the discharge time, m is the mass of active materials (g), and ΔV is the potential window of discharge. The cycling stability of the fibrous carbon mat and powder samples was carried out for over 1500 cycles.
Conclusions
Freestanding mats comprising carbon nanofibers were prepared successfully by using an abundant natural polymer, lignin, via electrospinning followed by carbonization at 900 °C. According to the morphology and pore analyses, the freestanding carbon mat exhibited a network structure consisting of carbon micro/nano fibers with high surface area, up to 2005 m2 g−1, as well as large pore volume, 0.7 cm3 g−1 with a dominant 70% mesopore content. The lignin-based carbon fibrous network afforded an excellent specific double layer capacitance, 205 F g−1, and maintained their capacity up to 83% over 1500 cycles. This outstanding electrochemical performance can be explained by the broad pore distribution in the mesopore range of the carbon fibers, which was induced by the combination of microphase separation between lignin and polyvinyl alcohol. A schematic representation of the microphase separation of the electrospun lignin/PVA fibers and the mesoporous structure in the carbon fiber is proposed (Fig. 1). This type of soft polymer templating allows the formation of mesoporous carbon nanofibers via electrospinning combined with chemical activation. Overall, we demonstrate that lignin can be easily converted into a high-end material for efficient energy storage technologies.Acknowledgements
This work was financially supported by the Academy of Finland through its Centres of Excellence Programme (2014–2019), HYBER and by the Cyber-Physical Microsystems project. The MOPPI project of the Aalto University Energy Efficiency (AEF) Program is also acknowledged. We thank the provision of facilities and technical support by Aalto University at OtaNano – Nanomicroscopy Center.References
- R. N. Reddy and R. G. Reddy, J. Power Sources, 2003, 124, 330 CrossRef CAS.
- Q. Huang, X. Wang and J. Li, Electrochim. Acta, 2006, 52, 1758 CrossRef CAS.
- K. Kierzek, E. Frackowiak, G. Lota, G. Gryglewicz and J. Machnikowski, Electrochim. Acta, 2004, 49, 515 CrossRef CAS.
- D. Qu and H. Shi, J. Power Sources, 1998, 74, 99 CrossRef CAS.
- E. Raymundo-Piñero, K. Kierzek, J. Machnikowski and F. Béguin, Carbon, 2006, 44, 2498 CrossRef.
- O. Barbieri, M. Hahn, A. Herzog and R. Kötz, Carbon, 2005, 43, 1303 CrossRef CAS.
- G. Salitra, A. Soffer, L. Eliad, Y. Cohen and D. Aurbach, J. Electrochem. Soc., 2000, 147, 2486 CrossRef CAS.
- B. Fang and L. Binder, J. Power Sources, 2006, 163, 616 CrossRef CAS.
- S. Shiraishi, H. Kurihara, K. Okabe, D. Hulicova and A. Oya, Electrochem. Commun., 2002, 4, 593 CrossRef CAS.
- J. N. Barisci, G. G. Wallace and R. H. Baughman, J. Electrochem. Soc., 2000, 147, 4580 CrossRef CAS.
- C. Portet, Z. Yang, Y. Korenblit, Y. Gogotsi, R. Mokaya and G. Yushin, J. Electrochem. Soc., 2009, 156, A1 CrossRef CAS.
- J. Lee, J. Kim and T. Hyeon, Adv. Mater., 2006, 18, 2073 CrossRef CAS.
- D. Saha, Y. Li, Z. Bi, J. Chen, J. K. Keum, D. K. Hensley, H. A. Grappe, H. M. Meyer, S. Dai, M. P. Paranthaman and A. K. Naskar, Langmuir, 2014, 30, 900 CrossRef CAS PubMed.
- B. Xu, F. Wu, R. Chen, G. Cao, S. Chen, Z. Zhou and Y. Yang, Electrochem. Commun., 2008, 10, 795 CrossRef CAS.
- Y. Zhang, H. Feng, X. Wu, L. Wang, A. Zhang, T. Xia, H. Dong, X. Li and L. Zhang, Int. J. Hydrogen Energy, 2009, 34, 4889 CrossRef CAS.
- B. E. Conway, Electrochemical Supercapacitors, Springer, US, 1999 Search PubMed.
- J. i. Hayashi, A. Kazehaya, K. Muroyama and A. P. Watkinson, Carbon, 2000, 38, 1873 CrossRef CAS.
- E. Gonzalez-Serrano, T. Cordero, J. Rodriguez-Mirasol, L. Cotoruelo and J. J. Rodriguez, Water Res., 2004, 38, 3043 CrossRef CAS PubMed.
- A. M. Dehkhoda, E. Gyenge and N. Ellis, Biomass Bioenergy, 2016, 87, 107 CrossRef CAS.
- K. Gergova, N. Petrov and S. Eser, Carbon, 1994, 32, 693 CrossRef CAS.
- O. N. Baklanova, G. V. Plaksin, V. A. Drozdov, V. K. Duplyakin, N. V. Chesnokov and B. N. Kuznetsov, Carbon, 2003, 41, 1793 CrossRef CAS.
- J. Rodriguez-Mirasol, T. Cordero and J. J. Rodriguez, Energy Fuels, 1993, 7, 133 CrossRef CAS.
- C. Liang, Z. Li and S. Dai, Angew. Chem., Int. Ed., 2008, 47, 3696 CrossRef CAS PubMed.
- E. Lim, C. Jo and J. Lee, Nanoscale, 2016, 8, 7827 RSC.
- J. Liu, T. Yang, D.-W. Wang, G. Q. Lu, D. Zhao and S. Z. Qiao, Nat. Commun., 2013, 4, 2798 Search PubMed.
- J. Xu, A. Wang and T. Zhang, Carbon, 2012, 50, 1807 CrossRef CAS.
- Y. Meng, D. Gu, F. Zhang, Y. Shi, L. Cheng, D. Feng, Z. Wu, Z. Chen, Y. Wan, A. Stein and D. Zhao, Chem. Mater., 2006, 18, 4447 CrossRef CAS.
- K. Yan, L.-B. Kong, Y.-H. Dai, M. Shi, K.-W. Shen, B. Hu, Y.-C. Luo and L. Kang, J. Mater. Chem. A, 2015, 3, 22781 CAS.
- D. A. Baker and T. G. Rials, J. Appl. Polym. Sci., 2013, 130, 713 CrossRef CAS.
- V. K. Thakur, M. K. Thakur, P. Raghavan and M. R. Kessler, ACS Sustainable Chem. Eng., 2014, 2, 1072 CrossRef CAS.
- A. J. Ragauskas, G. T. Beckham, M. J. Biddy, R. Chandra, F. Chen, M. F. Davis, B. H. Davison, R. A. Dixon, P. Gilna, M. Keller, P. Langan, A. K. Naskar, J. N. Saddler, T. J. Tschaplinski, G. A. Tuskan and C. E. Wyman, Science, 2014, 344, 6185 CrossRef PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552 CrossRef CAS PubMed.
- J. Fang, B. Yao, H. Nan, X. He and T. Wang, 2011.
- C. Lai, Z. Zhou, L. Zhang, X. Wang, Q. Zhou, Y. Zhao, Y. Wang, X.-F. Wu, Z. Zhu and H. Fong, J. Power Sources, 2014, 247, 134 CrossRef CAS.
- A. M. Navarro-Suarez, J. Carretero-Gonzalez, V. Roddatis, E. Goikolea, J. Segalini, E. Redondo, T. Rojo and R. Mysyk, RSC Adv., 2014, 4, 48336 RSC.
- J.-W. Jeon, L. Zhang, J. L. Lutkenhaus, D. D. Laskar, J. P. Lemmon, D. Choi, M. I. Nandasiri, A. Hashmi, J. Xu, R. K. Motkuri, C. A. Fernandez, J. Liu, M. P. Tucker, P. B. McGrail, B. Yang and S. K. Nune, ChemSusChem, 2015, 8, 428 CrossRef CAS PubMed.
- M. Ago, K. Okajima, J. E. Jakes, S. Park and O. J. Rojas, Biomacromolecules, 2012, 13, 918 CrossRef CAS PubMed.
- M. Ago, J. E. Jakes, L.-S. Johansson, S. Park and O. J. Rojas, ACS Appl. Mater. Interfaces, 2012, 4, 6849 CAS.
- Q. Cao, H.-s. Kim, N. Pimparkar, J. P. Kulkarni, C. Wang, M. Shim, K. Roy, M. A. Alam and J. A. Rogers, Nature, 2008, 454, 495 CrossRef CAS PubMed.
- G. H. Gelinck, H. E. A. Huitema, E. van Veenendaal, E. Cantatore, L. Schrijnemakers, J. B. P. H. van der Putten, T. C. T. Geuns, M. Beenhakkers, J. B. Giesbers, B.-H. Huisman, E. J. Meijer, E. M. Benito, F. J. Touwslager, A. W. Marsman, B. J. E. van Rens and D. M. de Leeuw, Nat. Mater., 2004, 3, 106 CrossRef CAS PubMed.
- D. J. Lipomi, B. C. K. Tee, M. Vosgueritchian and Z. Bao, Adv. Mater., 2011, 23, 1771 CrossRef CAS PubMed.
- D. J. Lipomi and Z. Bao, Energy Environ. Sci., 2011, 4, 3314 CAS.
- B. Shao, Q. Chen, R. Liu and L.-R. Zheng, Microw. Opt. Technol. Lett., 2012, 54, 226 CrossRef.
- S. Park, M. Vosguerichian and Z. Bao, Nanoscale, 2013, 5, 1727 RSC.
- N. Bhardwaj and S. C. Kundu, Biotechnol. Adv., 2010, 28, 325 CrossRef CAS PubMed.
- L. Persano, A. Camposeo, C. Tekmen and D. Pisignano, Macromol. Mater. Eng., 2013, 298, 504 CrossRef CAS.
- C.-L. Zhang and S.-H. Yu, Chem. Soc. Rev., 2014, 43, 4423 RSC.
- G. M. Irvine, Wood Sci. Technol., 1985, 19, 139–149 CrossRef CAS.
- J. F. Kadla, S. Kubo, R. A. Venditti, R. D. Gilbert, A. L. Compere and W. Griffith, Carbon, 2002, 40, 2913 CrossRef CAS.
- Y. Uraki, S. Kubo, N. Nigo, Y. Sano and T. Sasaya, Holzforschung, 1995, 49, 343 CrossRef CAS.
- M. A. Lillo-Ródenas, D. Cazorla-Amorós and A. Linares-Solano, Carbon, 2003, 41, 267 CrossRef.
- P. I. Ravikovitch and A. V. Neimark, J. Phys. Chem. B, 2001, 105, 6817 CrossRef CAS.
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774 RSC.
- J. Kastner, T. Pichler, H. Kuzmany, S. Curran, W. Blau, D. N. Weldon, M. Delamesiere, S. Draper and H. Zandbergen, Chem. Phys. Lett., 1994, 221, 53 CrossRef CAS.
- J. R. Kremer, D. N. Mastronasde and J. R. McIntosh, J. Struct. Biol., 1996, 116, 71 CrossRef CAS PubMed.
- P. Engelhardt, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons Ltd, Chichester, 2000, p. 4948 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra17536h |
| This journal is © The Royal Society of Chemistry 2016 |