
Squaric acid derivative effects on the kinetics of photopolymerization of different monomers
Janina Kabatc*,
Katarzyna Kostrzewska and
Katarzyna Jurek
UTP, University of Science and Technology, Faculty of Chemical Technology and Engineering, Seminaryjna 3, 85-326 Bydgoszcz, Poland. E-mail: nina@utp.edu.pl; Fax: +48 52 374 9005; Tel: +48 52 374 9112
First published on 15th July 2016
Abstract
Systems composed of 1,3-bis(phenylamino)squaraine (photosensitizer) and conventional free radical sources, such as tetramethylammonium n-butyltriphenylborate, diphenyliodonium chloride and diphenyliodonium hexafluorophosphate were used for initiation of photopolymerization occurring via a radical or cationic mechanism. The photopolymerization of 1,6-hexanediol diacrylate (HDDA), pentaerythritol triacrylate (PETA), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), cyclohexene oxide (CHO) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX) were carried out at 300 nm < λ < 500 nm irradiation. The polymerization kinetics was measured using a differential scanning calorimeter equipped with a high-pressure mercury lamp. The effect of co-initiator structure and type of monomer on the kinetics of the photopolymerization process is also presented here. It was found that the photoredox pairs, consisting of 1,3-bis(phenylamino)squaraine and tetramethylammonium n-butyltriphenylborate or diphenyliodonium salts, initiate radical and cationic polymerization in the UV-Vis light region. The photoinitiating ability of these new photoinitiating systems acting in the UV-Vis light region for initiation of polymerization of acrylates and epoxides was compared with a few commercially used photoinitiating systems.
1. Introduction
Typically polymerization is initiated by radical or ionic pathways but there are also several alternative examples of photopolymers that do not fit either of these mechanisms. Both radical and cationic polymerizations have been the basis for numerous conventional applications such as for coatings.1,2 Many technologically important monomers, such as vinyl ethers and oxiranes, are typically polymerized in cationic mode, alongside very important radically polymerizable monomers, such as mono- and multifunctional acrylates, and so the development of new more efficient radical and cationic initiators are desired.3–5 Photopolymerization by direct initiation by onium salts can be performed below 290 nm, but this limits their potential use in photopolymerization mainly when visible light-emitting sources are used.3 To overcome this problem, several indirect pathways, such as a combination of onium salts with photosensitizers, electron-donating or electron-accepting compounds have been extensively studied.6–9 For example, the excited state of tris(2,2′-bipyridine)ruthenium(II) reacts with an iodonium salt to generate a strong oxidant metal complex (Ru(bpy)33+) and phenyl radicals able to promote the formation of silyl radicals or silylium cations to initiate the polymerization.10 Other organometallic compounds have also been used as photosensitizers for different onium salts.11–13Even though different metal complexes have been introduced into PISs for polymer synthesis under very soft irradiation conditions such as sunlight, fluorescent bulbs or LED bulbs, these compounds are very expensive or difficult to prepare. Therefore, the development of organic dyes as substitutes with the advantages of lower cost, commercial availability, lower toxicity, better stability and solubility and easier extractability are clearly of interest.14 In the last decade, the following organic dyes: camphorquinone in dental composites,15 anthracene derivatives,16 pyrene derivatives,17 indanedione derivatives,18 N-substituted quinoxalinobenzothiazine derivatives,19 thiobarbituric acid derivative,20 chalcone derivatives,21 acridinedione derivatives,22 naphthalimide derivatives,23 diketopyrrolopyrrole-thiophene or diketopyrrolopyrrole-furan derivatives,24,25 violanthrone-7926 and other photosensitizers for onium salts have been used in dye mediated photoinitiating systems.27
As is seen, the use of free radical sources in combination with a suitable photosensitizer is a simple and flexible method to generate active species for cationic or radical polymerization. It is well known that polymerization can be carried out under a wide range of conditions, including variations in monomer structures, the number and type of reactive functional groups, temperature, atmosphere, source of energy (heat or light), irradiation rate and photoinitiator type.28
Generally, the following types of photoinitiators: free radical, cationic and anionic are known.28 There are also a few classes of initiators, which are able to initiate polymerization via both cationic and radical pathways, such as: iodonium and sulfonium salts, and arene complexes. For example, diaryliodonium salts have been very often applied as a source of protons, H+.29 The diaryliodonium salts are the most important photoinitiators because of their good thermal stability, solubility in most cationically polymerizable monomers, and their efficiency for generating cationic species during irradiation. UV-irradiation of iodonium salts leads to aryliodine radical cation formation, which reacts with solvent or monomer giving strong protic acid initiating polymerization of monomer.29
It should be noted that the development of efficient visible light emission sources, such as lasers and LEDs for imaging, printing and medical applications has increased the demand for initiator systems that are effective in the visible light region.30 Both alkyltriphenylborate and diphenyliodonium salts only absorb light below 300 nm, making them inactive for the applications where long-wavelength ultraviolet and visible light emission sources are used.30 Borate or diphenyliodonium salts with suitable sensitizers can initiate photopolymerization of different monomers using light with wavelength above 300 nm. Generally, there are two major strategies to extend the spectral sensitivity of borate and diphenyliodonium salts, photoreducible or photooxidizable sensitization.
New dye mediated photoinitiating systems are important for new and advanced applications, especially in the field of visible light curing. From this point of view, new squaraine-based sensitizers for iodonium and borate salts in cationic and free radical polymerization processes, are desirable. Yong He and co-workers studied two squaraine dyes, bis(1,2,3,3-tetramethylindolenium-2-ylidene)squaraine and bis(3-methylbenzothiazol-2-ylidene)squaraine, as sensitizers for three (p-octanoxyphenyl)phenyliodonium salts with varying counterion, in the radical polymerization of methyl methacrylate.31 The photoinitiating abilities of 2,2,3-trimethylindolenine-based squaraine dye incorporated in multicomponent systems for cationic polymerization of an epoxide or a vinyl ether as well as radical polymerization of TMPTA, have been investigated by Lalevée and co-workers.14,32 More generally, however, squarylium dyes are very rarely used as photosensitizers in photopolymerization processes. To our best knowledge, as yet, 1,3-bis(phenylamino)squaraine has not been studied in dye mediated photoinitiating systems. Moreover, this dye absorbs in the region from 350 to 450 nm and is thus amenable to blue light sources. In present paper, the photoinitiating ability of two-component systems composed of a squaric acid-based sensitizer and a variety of radical sources, tetramethylammonium n-butyltriphenylborate, diphenyliodonium chloride and diphenyliodonium hexafluorophosphate, in photopolymerization processes occurring via radical or cationic mechanisms, is described. The photoinitiating ability of the new photoinitiating systems, acting in the UV-Vis light region, for initiation of free radical polymerization of acrylates and epoxides was also compared with a few commercially used photoinitiators.
2. Experimental
2.1. Materials
Monomers (1,6-hexanediol diacrylate (HDDA), pentaerythritol triacrylate (PETA), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), cyclohexene oxide (CHO) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX)), co-initiators (diphenyliodonium chloride (I1) and diphenyliodonium hexafluorophosphate (I2)) and solvents (spectroscopic grade) were purchased from Aldrich (Poland) and used without further purification. 1,3-Bis(phenylamino)squaraine (SQ) and tetramethylammonium n-butyltriphenylborate (B2) were synthesized in our laboratory by methods described in the literature.33–352.2. Polymerization measurements
The kinetics of polymerization of all monomers photoinitiated by 1,3-bis(phenylamino)squaraine/tetramethylammonium n-butyltriphenylborate (SQ/B2), 1,3-bis(phenylamino)squaraine/diphenyliodonium chloride (SQ/I1) and 1,3-bis(phenylamino)squaraine/diphenyliodonium hexafluorophosphate (SQ/I2) was measured using a differential scanning calorimeter TA DSC Q2000 Instrument and TA Q PCA photo unit equipped with a high-pressure mercury lamp (Photo-DSC). The heat of the photoinitiated polymerization reaction, measured by means of a photo-differential scanning calorimeter, is a good control of the reaction temperature. UV-visible light (300 nm < λ < 500 nm) was applied from a high-pressure mercury lamp at a constant intensity of 30 mW cm−2 for several min under a nitrogen flow of 50 mL min−1 at a prescribed temperature (isothermal mode). A measured weight of the sample, 30 ± 0.1 mg, was placed into an open aluminum liquid DSC pan and the measurements were carried out under identical conditions. The sample was maintained at a prescribed temperature for 2 min before each measurement run began. Measurements were recorded at a sampling interval of 0.05 s per point. The polymerizing solution was composed of 1.8 mL of monomer, 0.2 mL of 1-methyl-2-pyrrolidinone and an appropriate amount of photoinitiator. The use of 1-methyl-2-pyrrolidinone was necessary due to the poor solubility of the dye in the monomers studied.The reaction heat liberated in the polymerization is directly proportional to the number of acrylates reacted in the system. By integrating the area under the exothermic peak, the conversion of the acrylate groups (C%) or the extent of the reaction was determined according to eqn (1):
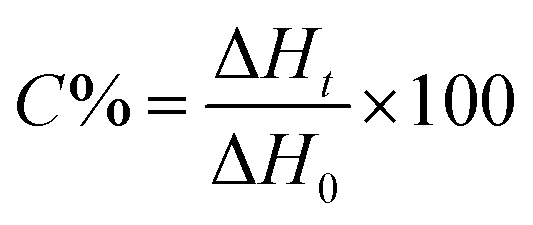 | (1) |
 | (2) |
2.3. Cyclic voltammetry measurements
The electrochemical measurements were evaluated by cyclic voltammetry (CV). Cyclic voltammetric measurements were made with an ER466 Integrated Potentiostat System (eDAQ, Poland) in a three-electrode configuration. The electrolyte was 0.1 M tetrabutylammonium perchlorate in dry acetonitrile. A 1 mm platinum wire electrode was applied as working electrode and platinum and Ag/AgCl were used as auxiliary and reference electrodes, respectively. All solutions were deoxygenated with N2 for at least 15 min prior to measurements. The computer-controlled potentiostat was equipped with EChem Software.3. Results and discussion
3.1. Excited state processes in dye mediated photoinitiating systems
An application of a suitable photosensitizer, that after excitation, can react with alkyltriphenylborate salt and diphenyliodonium salt via an electron transfer process gives the active species which can start the polymerization chain reaction. Fig. 1 schematically illustrates the reaction mechanism of the photoinitiating systems and photosensitizer structures studied. | ||
| Fig. 1 Schematic representation of the reaction mechanism of the photoinitiating systems and photosensitizer structures studied. | ||
In this strategy, the photoexcited sensitizer (squaraine dye, SQ*) is oxidized by diphenyliodonium salt (Ph2I+X−) to form the corresponding radical cation of sensitizer (SQ˙+) and diphenyliodonium radical (Ph2I˙). Next, the unstable diphenyliodonium radical undergoes irreversible rapid decomposition so as to prevent any reverse electron transfer and phenyl radical and iodobenzene are formed in this stage. It should be noted, that in the reaction occurring via this mechanism several cationic species are formed, which can also initiate the polymerization: the sensitizer-based radical cation (SQ˙+), which can start cationic polymerization, the strong protic acid formed from the reaction between the sensitizer-based radical cation (SQ˙+) and surrounding solvent molecules or monomer, as well as the carbocation of the monomer (M+), which is formed during the oxidation of the monomer radical (M˙) by diphenyliodonium salt.29,35–37 On the other hand, the photoexcited sensitizer (squaraine dye, SQ*) is reduced by alkyltriphenylborate salt (alkyl(Ph)3B−(CH3)4N+) to form sensitizer-based radical anion (SQ˙−) and boranyl radical alkyl(Ph)3B˙. In subsequent reaction the unstable boranyl radical undergoes C–B bond cleavage giving an alkyl radical (R˙) and stable triphenylboron. An alkyl radical can start the chain polymerization reaction. The mechanism of active species generation is shown in Scheme 1. As is shown in Scheme 1, for radically polymerizable monomers TMPTA, PETA and HDDA, the active species, such as butyl radical and phenyl radical are formed as a result of photoinduced electron transfer process between photoexcited sensitizer and an electron donor (borate salt) or an electron acceptor (iodonium salt). On the other hand, in the case of photopolymerization of cationically polymerizable monomers, the initiation species are formed from hydrogen abstraction of the monomer by the radical cation of squaraine dye and from the subsequent oxidation of the carbon-centered radicals of the monomer.
 | ||
| Scheme 1 Mechanism of generation of active species for the photopolymerization process. | ||
The main goal of the study was to apply a squaraine dye (1,3-bis(phenylamino)squaraine) as visible-light photosensitizer in dye mediated photoinitiating systems. This compound has a broad absorption band at approximately 400 nm (see Fig. 2) and can be applied as a visible sensitizer for the absorption of light above 350 nm. The half-width of the absorption spectrum is about 3000 cm−1. The molar extinction coefficient values are not high and range from 0.2 × 104 to 1.2 × 104 M−1 cm−1 over the illumination wavelengths used. The absorption band around 400 nm is due to the π→π* transition and the position of the absorption band depends on the polarity of the solvent.
 | ||
| Fig. 2 UV-Vis absorption spectra of squaraine in different solvents, recorded at room temperature. | ||
In order to confirm the electron transfer between n-butyltriphenylborate salt (B2) or iodonium salts (I1, I2) and the dye, fluorescence quenching experiments were performed at different concentrations of co-initiators. The structure of the co-initiators are presented in Fig. 3.
 | ||
| Fig. 3 Structures and abbreviation of co-initiators studied. | ||
For this purpose, the study of changes in both fluorescence intensity and fluorescence lifetime under increasing concentration of quencher (co-initiator) was studied.37,38 The influence of quenchers on the fluorescence intensities and fluorescence lifetimes is presented in Fig. 4 (I2) and Table 1 (B2, I1, I2), respectively.
 | ||
| Fig. 4 The effect of diphenyliodonium hexafluorophosphate (I2) on the fluorescence intensity of 1,3-bis(phenylamino)squaraine in 1-methyl-2-pyrrolidinone as solvent; λEX = 370 nm, and λEM = 440 nm. | ||
| Co-initiator concentration [M] | Fluorescence lifetime | ||
|---|---|---|---|
| B2 [ns] | I1 [ns] | I2 [ns] | |
| a λEX = 370 nm, λEM = 440 nm. | |||
| 0 | 8.0552 | 8.0552 | 8.0552 |
| 1.0 × 10−4 | 6.6514 | 7.5734 | 4.7261 |
| 2.0 × 10−4 | 6.2689 | 7.2019 | 4.62 |
| 3.0 × 10−4 | 6.0698 | 6.9457 | 4.5713 |
| 4.0 × 10−4 | 5.5764 | 6.7155 | 4.5018 |
| 5.0 × 10−4 | 5.5571 | 6.464 | 4.4241 |
| 6.0 × 10−4 | 5.3186 | 6.2186 | 4.2558 |
| 7.0 × 10−4 | 5.236 | 5.9575 | 4.2837 |
| 8.0 × 10−4 | 5.0116 | 5.7081 | 4.2115 |
| 9.0 × 10−4 | 4.9441 | 5.5116 | 4.1566 |
| 1.0 × 10−3 | 4.9263 | 5.3816 | 4.0706 |
| 1.5 × 10−3 | 4.6455 | 5.0397 | 3.8678 |
| 2.0 × 10−3 | 4.2855 | 4.6269 | 3.6827 |
| 3.0 × 10−3 | 3.7565 | 3.8739 | 3.3825 |
| 4.0 × 10−3 | 3.4134 | 3.3821 | 3.0513 |
| 5.0 × 10−3 | 3.1794 | 3.1321 | 2.9366 |
An addition of borate salt or iodonium salt results in a significant decrease of the fluorescence intensity and decrease of lifetime of the excited singlet state of the dye. Moreover, the absence of any new peaks in the emission spectra excludes any exciplex formation.
For calculation of the bimolecular quenching rate constants kq, from e.g. (3), the fluorescence lifetime of dye (τ0) without any quencher was used:
 | (3) |
Taking into account the fluorescence lifetime τ0, and the slope of the linear relationship of Stern–Volmer plot, the kq value may be calculated.
As was mentioned in our previous paper39 the rate of dynamic quenching of the excited singlet state is different for borate salt in comparison with iodonium salts. The quenching rate constants calculated for low concentrations of borate salt differs from that for higher concentrations of quencher. The slopes of the Stern–Volmer linear relationship observed are as follows: 522.10 (B2, <1 × 10−3 M), 231.39 (B2, >1 × 10−3 M), 318.52 (I1) and 150.31 (I2). From the Stern–Volmer equation plot, the corresponding rate constants of fluorescence quenching reaction were 6.48 × 1010, 2.87 × 1010 , 3.95 × 1010 and 1.87 × 1010 M−1 s−1, which are nearly one order of magnitude larger than that of diffusion controlled bimolecular reaction constant (∼2 × 109 M−1 s−1). These results confirmed that the onium salts under study are very effective fluorescence quenchers for excited (SQ) dye and the fast quenching occurs predominantly through the intramolecular ion-pair pathway.
The quenching of the excited singlet state of the dye by borate salt is much more efficient in the low concentration range of borate salt. The rate constant of quenching by diphenyliodonium hexafluorophosphate is lower than that observed for diphenyliodonium chloride. This is probably due to a larger counter ion in salt (I2) than in diphenyliodonium chloride (I1), which reduces the mobility of the salt (I2).
The influence of borate salt and iodonium salts on the rate of fluorescence decay of the dye suggests that the primary photoreaction occurs between the dye and borate salt and both iodonium salts. This phenomenon may be a result of photoinduced electron transfer process occurring between the excited dye molecule and quencher in the ground state. During the photoinduced electron-transfer process squaraine may act as an electron donor or an electron acceptor depending on the electrochemical properties of both dye and quencher.
In conclusion, owing to the energy of the excited (SQ) dye being lower than that of borate and both diphenyliodonium salts (co-initiators), energy transfer from excited (SQ) to co-initiator is not possible. Therefore, it is reasonable to consider that upon irradiation the photoinduced electron transfer reaction between excited squaraine dye and onium salts occurs via an intramolecular pathway, which has a large reaction rate as shown by the fluorescence quenching experiments described above.
Additionally, the mechanism of primary and secondary reactions occurring during irradiation of the photoinitiating systems under study may be in part supported by cyclic voltammetry experiments. In order to estimate thermodynamically the activity of the squaraine dye/borate salt and squaraine dye/iodonium salt photoreaction, the values of free energy change (ΔGel) for the electron transfer reaction were calculated according to the Rehm–Weller equation.40
| ΔGel = Eox(D˙+/D) − Ered(A/A˙−) − Ze2/εa − E00 | (4) |
3.2. Effect of active species source (radicals, cations) on photopolymerization
The polymerization of different monomers, such as 1,6-hexanediol diacrylate (HDDA), pentaerythritol triacrylate (PETA), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), cyclohexene oxide (CHO) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX) with 5 × 10−3 M of photosensitizer (squaraine dye) was performed to examine the efficiency of three different onium salts (co-initiators), tetramethylammonium n-butyltriphenylborate (B2), diphenyliodonium chloride (I1) and diphenyliodonium hexafluorophosphate (I2) in radical polymerization of HDDA, PETA and TMPTA, as well as in cationic polymerization of CHO and EPOX in the presence and absence of active co-initiator species (5 × 10−3 M). The photopolymerizations were conducted using light wavelengths in the range from 300–500 nm and an irradiation intensity of 30 mW cm−2. The structure of the monomers used are shown in Fig. 5. | ||
| Fig. 5 The structures of monomers used. | ||
In absence of active species source (co-initiator), the photopolymerization of all monomers studied was carried out with 1,3-bis(phenylamino)squaraine (SQ) (5 × 10−3 M) at irradiation in the range 300–500 nm and no monomer conversion was observed. Fig. 6–8 show the kinetic curves recorded during photopolymerization of radically polymerizable monomers, 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), pentaerythritol triacrylate (PETA) and 1,6-hexanediol diacrylate (HDDA), as well as the degree of double bond conversion as a function of irradiation time. Fig. 9 presents the kinetic and time–conversion curves recorded during cationic polymerization of cyclohexene oxide (CHO) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX) initiated by the system composed of 1,3-bis(phenylamino)squaraine and diphenyliodonium hexafluorophosphate. It can be seen that with increasing of irradiation time, the polymerization proceeds and attains limiting conversions with each photosensitizer/radical source combination.
 | ||
| Fig. 6 The kinetic and time–conversion curves recorded during radical polymerization of TMPTA initiated by 1,3-bis(phenylamino)squaraine in the presence of radical sources (marked in the figure) at ambient temperature; [SQ] = 5 × 10−3 mol L−1; [co-initiator] = 5 × 10−3 mol L−1. Light intensity was equal 30 mW cm−2. | ||
 | ||
| Fig. 7 The kinetic and time–conversion curves recorded during radical polymerization of PETA initiated by 1,3-bis(phenylamino)squaraine in presence of radical sources (marked in the figure) at ambient temperature; [SQ] = 5 × 10−3 mol L−1; [co-initiator] = 5 × 10−3 mol L−1. Light intensity was equal 30 mW cm−2. | ||
 | ||
| Fig. 8 The kinetic and time–conversion curves recorded during radical polymerization of HEDA initiated by 1,3-bis(phenylamino)squaraine in presence of radical sources (marked in the figure) at ambient temperature; [SQ] = 5 × 10−3 mol L−1; [co-initiator] = 5 × 10−3 mol L−1. Light intensity was equal 30 mW cm−2. | ||
 | ||
| Fig. 9 The kinetic and time–conversion curves recorded during cationic polymerization of cyclohexene oxide (CHO) and 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX) initiated by the pair composed of 1,3-bis(phenylamino)squaraine and diphenyliodonium hexafluorophosphate (I2) at ambient temperature; [SQ] = 5 × 10−3 mol L−1; [co-initiator] = 5 × 10−3 mol L−1. Light intensity was equal 30 mW cm−2. | ||
Preliminary experiments showed that each monomer used did not polymerize at irradiation (300–500 nm) if any one component of the photoinitiator system was missing. The data presented in Fig. 6–9 and Table 2 clearly indicate that the efficiency of monomer polymerization depends on both the type of co-initiator and monomer used.
| Co-initiator | Heat flow [mW] | Rp [mmol s−1] | Total monomer conversion [%] |
|---|---|---|---|
| a The concentration of squaraine dye and co-initiator was equal to 5 × 10−3 mol L−1 and light intensity was 30 mW cm−2. | |||
| TMPTA | |||
| B2 | 16.70 | 0.66 | 10.5 |
| I1 | 64.2 | 2.53 | 17.5 |
| I2 | 20.78 | 0.82 | 6.9 |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
|||
| PETA | |||
| B2 | 49.43 | 1.96 | 21.6 |
| I1 | 12.98 | 0.52 | 19.1 |
| I2 | 12.57 | 0.50 | 10 |
|
|||
| HDDA | |||
| B2 | 2.14 | 0.054 | 27.32 |
| I1 | 5.06 | 0.13 | 1.0 |
| I2 | 4.39 | 0.11 | 6.8 |
|
|||
| CHO | |||
| I2 | 28.75 | 0.97 | 69.5 |
The highest efficiency for pentaerythritol triacrylate (PETA) radical polymerization was observed in the case of tetramethylammonium n-butyltriphenylborate used as co-initiator. The radical polymerization of PETA occurs with the degree of double bond conversion two-times higher for n-butyltriphenylborate salt than for diphenyliodonium hexafluorophosphate. The highest efficiency of 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA) radical polymerization was observed for diphenyliodonium salt (I1), whereas the lowest efficiency was obtained for diphenyliodonium salt with hexafluorophosphate counter-ion (I2). The maximum conversion of TMPTA obtained during the polymerization initiated by squaraine dye in the presence of B2, I1 and I2 was found to be 10.5, 17.5 and 6.9, respectively. The order of initiator activity was observed as I1 > B2 > I2. This trend can be explained by the difference in the decomposition rate constant and the reactivity of active species formed toward the functional group of monomer.
In the case of the two-functional group monomer 1,6-hexanediol diacrylate (HDDA), the highest value of double bond conversion was observed for polymerization initiated by borate salt (B2). The results obtained may be related to the different properties of monomers used with the rate of radical polymerization ranging from 0.11 to 2.53 mmol s−1. The highest rates of radical polymerization were achieved for three-functional group monomers: 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate and pentaerythritol triacrylate. The radical polymerization of diacrylate occurs with the lowest rates for all co-initiators used. As shown in Scheme 1, the polymerization of radically polymerizable monomers is initiated by butyl and phenyl radicals. The differences in photoinitiation ability observed between diphenyliodonium salts used may be related to different reduction potentials of the iodonium salts, resulting in different values of the free energy change for electron transfer process.
From the data presented in Table 2 it is seen that the conversion of radically polymerizable monomers such as triacrylates, achieved during the photopolymerization process initiated by the system composed of squaraine (SQ) and diphenyliodonium chloride, was about 20% for 10 min of irradiation. Therefore, diphenyliodonium chloride is a relatively more efficient radical source than the others to accelerate the rate of polymerization. The higher activity of diphenyliodonium chloride can be explained by high efficiency of radical formation.3,37,38 The kinetic results obtained for the radically polymerizable monomers shown that the systems under study initiate polymerization process faster than other squaraine-based photoinitiators. For example, for the two-component photoinitiating systems composed of bis(1,2,3,3-tetramethylindolenium-2-ylidene)squaraine or bis(3-methylbenzothiazol-2-ylidene)squaraine, and (p-octanoxyphenyl)phenyliodonium salt, the maximum double bond conversion of methyl methacrylate are in the range of 10–14% after 4 h of irradiation time.31 However, photopolymerization of TMPTA in laminate initiated by 1,3,3-trimethylindolenine-based squaraine dye and diphenyliodonium chloride (0.5%/2%, w/w) leads to a conversion of monomer of about 40%.32 When camphorquinone and diphenyliodonium chloride (0.5%/2%, w/w) was used to initiate TMPTA polymerization under a halogen lamp as light source, final monomer conversion obtained in laminate was about 18%.41
On the other hand, the photopolymerization of cationically polymerizable monomer, such as 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate (EPOX) does not occur, or does so only at a very low rate. In the case of polymerization of cyclohexene oxide (CHO) only one of the studied photoinitiating systems, composed of squaraine dye and diphenyliodonium hexafluorophosphate (I2), initiates photopolymerization. In this case the initiating species are formed as a result of primary photochemical processes between the photoexcited sensitizer and diphenyliodonium salt. Total monomer conversion is about 70% after 60 min of irradiation time. It is known, that in photopolymerization of cyclohexene oxide (CHO), the monomer plays a role of hydrogen donor.29,36 Therefore, photosensitizer-based radical cations may abstract hydrogen from cyclohexene oxide (CHO) to form two different cations (Scheme 2).
 | ||
| Scheme 2 The primary and secondary processes between photosensitizer, electron donor and proton donor (for example, monomer). | ||
In route A sensitizer-based radical cation (SQ˙+) reacts with cyclohexene oxide (CHO) via a hydrogen abstraction. Then it donates a proton to regenerate photosensitizer (dye). A similar mechanism as a crucial step in the mechanism of cationic polymerization of cyclohexene oxide (CHO) initiated by naphthoylenebenzimidazolone dyes and iodonium salt was proposed by Podsiadły et al.30 Based on the results described by Podsiadły et al. the formation of cation via the second reaction route is suggested to occur. In the next step, the abstraction of the epoxide hydrogen by sensitizer-based radical cation (SQ˙+) or phenyl radical (Ph˙) (Schemes 1 and 2) results in carbon-centered cyclohexene oxide radicals (monomer radicals), which should form readily because the radicals are stabilized by resonance. However, the monomer radical may also be oxidized by diphenyliodonium salt to yield a carbocation. In a subsequent step, diphenyliodonium radical undergoes fast and irreversible decomposition to iodobenzene and phenyl radical. The phenyl radical may also abstract a hydrogen atom from the monomer.30,36–38
It has been empirically noted, for example by Crivello,42,42 Sinka44 and Lapin45 that certain epoxide monomers display high reactivity in photoinitiated cationic polymerization and are suitable for such use while others undergo apparently sluggish reactions and are not. Still other epoxy monomers that possess basic groups as part of their structures are not polymerizable under cationic conditions.42 For this reason, the range of currently available epoxide monomers, that are useful in applications involving photoinitiated cationic polymerization is limited. Crivello43 explained the reasons for the reactivity differences that have been observed between various epoxide monomers. He reported that the different characteristic types of epoxide monomers kinetic behaviors in cationic polymerization can be studied by measurement of their conversion vs. irradiation time.46 Based on this, the kinetic results presented in Fig. 9 show the responses of these two epoxide monomers and are characteristic of two extremes in the photopolymerization behavior exhibited by these monomers under cationic ring-opening polymerization conditions.42 Cyclohexene oxide (CHO) is representative of what can be termed a “class I” monomer that undergoes very rapid polymerization on UV irradiation with very little induction period. The generally accepted mechanism for the photoinitiated cationic ring-opening polymerization of heterocyclic ether monomers, that includes epoxides and diaryliodonium salts as photoinitiators, occurs via four steps.42,47 Irradiation of photoinitiator results in excitation and then fragmentation into a variety of radical and cationic species. Further reaction of those species with monomer, solvent or other protogenic components present yields the Brønsted acid HX. The photogenerated superacid very rapidly protonates the cyclic ether and the secondary oxonium species are formed. In a subsequent process, the secondary oxonium species undergo SN2 attack by the nucleophilic cyclic ether monomer to yield the tertiary oxonium species as a result of ring-opening of the heterocyclic ring.42,48
Finally, repetitive attack by a cyclic ether monomer on the tertiary oxonium ion leads to polymer chain growth. Generally, it is known that the rate of tertiary oxonium species formation is the limiting rate of polymerization process. Based on the Crivello studies, it was concluded that the overall rate of polymerization depends on the type of monomer undergoing polymerization.42 Cyclohexene oxide (CHO) is a highly strained monomer and possesses no structural or electronic features that can stabilize the secondary oxonium ion, therefore the activation energy for the nucleophilic cyclohexene oxide (CHO) attack on the secondary oxonium ion leading to the formation of corresponding tertiary oxonium ion is relatively small, and is thermally accessible at room temperature.42 With such highly reactive monomer, such as cyclohexene oxide (CHO), the overall rate of the polymerization depends on the rate of the photolysis of photoinitiator which delivers the highly reactive protonic acid initiator to the system. The kinetic results obtained for the photoinitiating system composed of squaraine dye and diphenyliodonium hexafluorophosphate are similar to those obtained by Crivello et al. for the system poly(N-vinylcarbazole)/(4-n-decyloxyphenyl)phenyliodonium hexafluoroantimonate.49
In contrast, under the same conditions, 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate, a typical “class III” monomer, undergoes very slow photopolymerization.19 Such type of monomers possess oxygen atoms that are less effective in the stabilizing of the intermediates. These monomers undergo slow, retarded photoinitiated cationic polymerization without a pronounced induction period.
Taking this into account, one can conclude that this is the reason for the different kinetic behavior of two epoxide monomers studied.
It should be also noted that the results obtained from fluorescence quenching experiment are consistent with the kinetics results. The highest value of fluorescence quenching rate constant was observed for diphenyliodonium chloride used as a quencher. Therefore, the rate of active species formation (radicals) in this photoredox pair is highest and the initiation of polymerization occurs faster.
In the next step, to fully demonstrate the usefulness of the proposed approach, the photoinitiating ability was compared to commercially available photoinitiators. It is well known that the photoinitiating ability is highly dependent on the nature of the light source, its intensity and wavelength, as well as on reactivity of the formulation.
For example, the photopolymerization of an acrylate monomer, trimethylolpropane triacrylate (TMPTA) in the presence of phosphine oxide as the photoinitiator leads to 5% of double bond conversion under air and irradiation with an Xe lamp.50 Addition of 3% w/w of tris(trimethylsilyl)silane causes in an increase of conversion to about 40% under the same conditions.50 Monoacylphosphine oxides such as Speedcure TPO initiate radical polymerization of TMPTA under irradiation with LED at 400 nm and light intensity equal to 85 mW cm−2. The maximum double bond conversion was about 15%.51 Arsu and co-workers52 showed that the polymerization of TMPTA using BAPO (0.2 wt%) in the absence and presence of 2-mercaptothioxanthone (0.02 wt%), during irradiation with a medium-pressure mercury lamp with a light intensity of 20 mW cm−2, occurs with conversions of 30 and 38%, respectively.52 Shi et al. in 2009 described a series of benzophenone-terminated hyperbranched polyesters bearing amine moieties as photoinitiator for radical polymerization of trimethylolpropane triacrylate. The maximal conversion achieved was in range from 44 to 59%.53 Yang and co-workers studied the kinetics of radical polymerization using a UV-lamp (200–400 nm) as a light source with an intensity of 20 mW cm−2. The dibenzoyl peroxide does not initiate polymerization of TMPTA under these conditions. However, application of isopropyl thioxanthone or thioxanthone-based N-phthalimidoamino acid ammonium salt as a photoinitiator gives a final conversion of about 40 and 83%, respectively.54 Arsu and co-workers studied an amine linked benzophenone photoinitiator for free radical polymerization of triacrylates.52 A medium-pressure mercury arc lamp (220–400 nm) with a light intensity of 40 mW cm−2 was used as the light source. The final conversion changes were in the range from 12 to 55%.
On the basis of the studies of Lalevée and co-workers 54,55 on the application of different core-pyrene compounds in the presence of N-methyldiethanolamine (MEDA), 2-bromoacetophenone, N-vinylcarbazole and diphenyliodonium salt initiate both cationic polymerization of 3,4-epoxycyclohexanemethyl 3,4-epoxycyclohexylcarboxylate (EPOX) and radical polymerization of trimethylolpropane triacrylate (TMPTA) upon exposure to an Xe–Hg lamp under air, with conversion from 5 to 70% and from 5 to 55% form EPOX and TMPTA, respectively.54
An epoxy monomer ((3,4-epoxycyclohexane)methyl 3,4-epoxycyclohexylcarboxylate (EPOX)) in the presence of commercially available components of a photoinitiating system, such as hydroxy alkyl acetophenone and diphenyliodonium salt (1%/1% w/w) does not polymerize under air and Xe lamp exposure, as well as in presence of 2% w/w of iodonium salt under air upon 365 nm LED exposure (∼50 mW cm−2). However, an addition of 3% (w/w) of tris(trimethylsilyl)silane gives 60% conversion of epoxide.16,50 The polymerization of epoxide under air upon xenon lamp exposure (60 mW cm−2) in the presence of bis(acyl)phosphine oxide (BAPO) and diphenyliodonium salt (1%/1% w/w) gives 30% conversion of monomer.16,50 Ruthenium complexes have been studied as photosensitizers in radical polymerization under soft visible irradiation. EPOX under air upon halogen lamp irradiation (12 mW cm−2) in the presence of BAPO and diphenyliodonium chloride (1%/2% w/w) or N-vinylcarbazole and diphenyliodonium salt (1%/2% w/w) undergoes cationic polymerization. The conversion of monomer is equal to 2 and 10%, respectively.16,50,55–59
For comparison, the total conversion obtained for photoinitiating systems in our study are about 18, 22, 27 and 70% for TMPTA, PETA, HDDA and CHO, respectively.
4. Conclusions
Photoredox pairs, consisting of 1,3-bis(phenylamino)squaraine and tetramethylammonium n-butyltriphenylborate salt or commercially available diphenyliodonium salts, may be used as ultraviolet-visible wavelengths initiators for the radical polymerization of pentaerythritol triacrylate (PETA), 2-ethyl-2-(hydroxymethyl)-1,3-propanediol triacrylate (TMPTA), 1,6-hexanediol diacrylate (HDDA), as well as cationic polymerization of cyclohexene oxide (CHO). It is assumed that the photochemical decomposition of co-initiator molecule results in formation of active species that further initiate polymerization. The order of initiator activity was observed as I1 > B2 > I2. This trend can be explained by the difference in the decomposition rate constant and the reactivity of active species formed toward the functional group of monomer. In such dye-based photoinitiating systems, the initiating species are typically generated by photoinduced electron transfer processes. The fluorescence quenching rate constants are close to the rate of diffusion-controlled reaction. The fluorescence quenching process influences the rate of photopolymerization process with an increase in the fluorescence quenching rate constant resulting in an increase of the rate of initiation of the polymerization process. Moreover, the efficiency of polymerization depends on both the type of co-initiator and monomer used. The highest efficiency for pentaerythritol triacrylate (PETA) radical polymerization was observed in the case of borate salt used as co-initiator. In the case of cationic polymerization, the highest efficiency was obtained for cyclohexene oxide (CHO) when diphenyliodonium hexafluorophosphate was used as a co-initiator with monomer conversion of about 70% after 10 min of irradiation.In the case of photopolymerization of cationically polymerizable monomers, the initiation species are formed from the hydrogen abstraction of the monomer by the cationic radicals of squaraine dye and from the subsequent oxidations of the carbon-centered radicals of the monomer.
The novelty of this manuscript is the introduction of squaraine dye chemistry, which opens a new way to cure coatings under UV and visible light. Interestingly, this also allows photocuring under soft conditions (visible light using irradiation of Xe or Hg lamps), diode lasers (405, 432 nm, LED bulbs; low intensity sources) under air and using relatively low viscosity monomers.
Acknowledgements
This work was supported by The National Science Centre (NCN) (Cracow, Poland), Grant No. 2013/11/B/ST5/01281.References
- Z.-S. Wang and F. Liu, Front. Chem. China, 2010, 5, 150–161 CrossRef
.
- P. V. Kamat, S. Das, K. G. Thomas and M. V. George, J. Phys. Chem., 1992, 96, 195–199 CrossRef CAS
- M. K. Gupta and R. P. Singh, Polym. Bull., 2009, 62, 271–280 CrossRef CAS
- J. V. Crivello, Adv. Polym. Sci., 1984, 1, 62 Search PubMed
- M. K. Grupta and R. P. Singh, Polym. Bull., 2008, 60, 755 CrossRef
- Y. Yagci and W. Schnabel, Makromol. Chem., Macromol. Symp., 1992, 60, 133 CrossRef CAS
- F. A. M. Rasoul Abdul, A. Ledwith and Y. Yagci, Polymer, 1978, 19, 1219 CrossRef
- Y. Chen, T. Yamamura and K. Igarashi, J. Polym. Sci., Part A: Polym. Chem., 2000, 38, 90 CrossRef CAS
- D. Dossow, Q. Zhu, G. Hizal, Y. Yagci and W. Schnabel, Polymer, 1996, 37, 2821 CrossRef CAS
- J. Lalevée, N. Blanchard, M. A. Tehfe, F. Morlet-Savary and J. P. Fouassier, Macromolecules, 2010, 43, 10191–10195 CrossRef
- D. L. Versace, F. Dalmas, J. P. Fouassier and J. Lalevée, ACS Macro Lett., 2013, 2, 341–345 CrossRef CAS
- M. A. Tehfe, F. Dumur, S. Telitel, D. Gigmes, E. Contal, D. Bertin, F. Morlet-Savary, B. Graff, J. P. Fouassier and J. Lalevée, Eur. Polym. J., 2013, 49, 1040–1049 CrossRef CAS
- W. Wu, P. Yang, L. Ma, J. Lalevée and J. Zhao, Eur. J. Inorg. Chem., 2013, 228–231 CrossRef CAS
- P. Xiao, J. Zhanga, F. Dumur, M. A. Tehfe, F. Morlet-Savary, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Prog. Polym. Sci., 2015, 41, 32–66 CrossRef CAS
- A. Vitale, M. Sangermano, R. Bongiovanni, P. Burtscher and N. Moszner, Materials, 2014, 7, 554–562 CrossRef
- M. A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff, N. Blanchard and J. P. Fouassier, ACS Macro Lett., 2012, 1, 198–203 CrossRef CAS
- M. A. Tehfe, F. Dumur, E. Contal, B. Graff, F. Morlet-Savary, D. Gigmes, J. P. Fouassier and J. Lalevée, Polym. Chem., 2013, 4, 1625–1634 RSC
- M. A. Tehfe, F. Dumur, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 3332–3341 CrossRef CAS
- R. Podsiadły and R. Strzelczyk, Dyes Pigm., 2013, 97, 462–468 CrossRef
- M. A. Tehfe, F. Dumur, B. Graff, F. Morlet-Savary, D. Gigmes, J. P. Fouassier and J. Lalevée, Polym. Chem., 2013, 4, 3866–3875 RSC
- M. A. Tehfe, F. Dumur, P. Xiao, M. Delgove, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2014, 5, 382–390 RSC
- P. Xiao, F. Dumur, M. A. Tehfe, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromol. Chem. Phys., 2013, 214, 2276–2282 CrossRef CAS
- P. Xiao, F. Dumur, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2014, 47, 601–608 CrossRef CAS
- P. Xiao, W. Hong, Y. Li, F. Dumur, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Polymer, 2014, 55, 746–751 CrossRef CAS
- P. Xiao, W. Hong, Y. Li, F. Dumur, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Polym. Chem., 2014, 5, 2293–2300 RSC
- M. A. Tehfe, D. Gigmes, F. Dumur, D. Bertin, F. Morlet-Savary, B. Graff, J. Lalevée and J. P. Fouassier, Polym. Chem., 2012, 3, 1899–1902 RSC
- S. Dadashi-Silab, S. Doran and Y. Yagci, Chem. Rev., 2016 DOI:10.1021/acs.chemrev.5b00586
- E. Andrzejewska, Prog. Polym. Sci., 2001, 26, 605–665 CrossRef CAS
- W. Schnabel, in Polymers and light. Fundamentals and technical application, Wiley-VCH, Weinheim, 2007, pp. 275–329 Search PubMed
- R. Podsiadły, A. Maruszewska, R. Michalski, A. Marcinek and J. Kolińska, Dyes Pigm., 2012, 95, 252–259 CrossRef
- Y. He, W. Zhou, F. Wu, M. Li and E. Wang, J. Photochem. Photobiol., A, 2004, 162, 463–471 CrossRef CAS
- P. Xiao, F. Dumur, T. T. Bui, F. Goubard, B. Graff, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, ACS Macro Lett., 2013, 2, 736–740 CrossRef CAS
- R. Daminco, J. Org. Chem., 1964, 29, 1971–1976 CrossRef
- S.-Y. Park, K. Jun and S.-W. Oh, Bull. Korean Chem. Soc., 2005, 26, 428–432 CrossRef CAS
- H. Junek, A. Hermetter, H. F. Colbrie and H. Aigner, Tetrahedron Lett., 1973, 14, 2993–2996 CrossRef
- J. V. Crivello and M. Sangermano, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 343–356 CrossRef CAS
- Y. Yagci, S. Jockusch and N. J. Turro, Macromolecules, 2010, 43, 6245–6260 CrossRef CAS
- S. Deniizligil, Y. Yagci and C. MacArdle, Polymer, 1995, 36, 3093 CrossRef
- J. Kabatc, K. Kostrzewska, K. Jurek, R. Dobosz and Ł. Orzeł, Dyes Pigm., 2016, 127, 179–186 CrossRef CAS
- D. Rehm and A. Weller, Isr. J. Chem., 1970, 8, 259 CrossRef CAS
- X. Allonas, J. P. Fouassier, L. Angiolini and D. Caretti, Helv. Chim. Acta, 2001, 84, 2577–2588 CrossRef CAS
- U. Bulut and J. V. Crivello, Macromolecules, 2005, 38, 3584–3595 CrossRef CAS
- J. V. Crivello, J. H. W. Lam and C. N. Volante, J. Radiat. Curing, 1977, 4, 2 CAS
- J. V. Sinka and D. Mazzoni, in Proc. Radtech 88 North America Conf., New Orleans, LA, 24–28 April 1988, pp. 378–388 Search PubMed
- S. C. Lapin, Radiation curing of polymeric materials, in ACS Symposium Series No. 417, ed. C. E. Hoyle and J. F. Kinstle, Americal Chemical Society, Washington, DC, 1990, pp. 361–381 Search PubMed
- J. V. Crivello, Des. Monomers Polym., 2002, 5, 141 CrossRef CAS
- S. Penczek, P. Kubisa and K. Matyjaszewski, Adv. Polym. Sci., 1980, 37, 11 Search PubMed
- N. Corrigan, S. Shanmugam, J. Xu and C. Boyer, Chem. Soc. Rev., 2016 10.1039/c6cs00185
- K. Sampson, A. Paik, B. Duval and D. D. L. Whalen, J. Org. Chem., 2004, 69, 5204–5211 CrossRef CAS PubMed
- Y. Hua and J. V. Crivello, Macromolecules, 2001, 34, 2488–2494 CrossRef CAS
- M. A. Tehfe, F. Louradour, J. Lalevée and J. P. Fouassier, Appl. Sci., 2013, 3, 490–514 CrossRef CAS
- S. Keskin, S. Jockusch, N. J. Turro and N. Arsu, Macromolecules, 2008, 41, 4631–4634 CrossRef CAS
- L. Hu, A. Asif, J. Xie and W. Shi, Polym. Adv. Technol., 2011, 22, 1673–1680 CrossRef CAS
- M. A. Tehfe, J. Lalevée, S. Telitel, E. Contal, F. Dumur, D. Gigmes, D. Bertin, M. Nechab, M. Bertrand and F. Morlet-Savary, Macromolecules, 2012, 45, 4454–4460 CrossRef CAS
- M. A. Tehfe, J. Lalevée, F. Morlet-Savary, B. Graff and J. P. Fouassier, Macromolecules, 2011, 44, 8374–8379 CrossRef CAS
- P. Sehnal, K. Harper, A. T. Rose, D. G. Anderson, W. A. Green, B. Husár, M. Griesser and R. Liska, Novel Phosphine Oxide Photoinitiators, RadTech, 2014 Search PubMed
- M.-H. He, R.-X. Xu, G.-X. Chen, Z.-H. Zeng and J.-W. Yang, Chin. Chem. Lett., 2014, 25, 1445–1448 CrossRef CAS
- S. Telitel, F. Dumur, T. Faury, B. Graff, M. A. Tehfe, D. Gigmes and J. P. Fouassier, Beilstein J. Org. Chem., 2013, 9, 877–890 CrossRef CAS PubMed
- G. Temel, B. Enginol, M. Aydin, D. K. Balta and N. Arsu, J. Photochem. Photobiol., A, 2011, 219, 26–31 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2016 |