
Biodegradable thermal- and redox-responsive poly(L-glutamate) with Y-shaped oligo(ethylene glycol) side-chain and tunable phase transition temperature†
Xiaohui Fuab,
Yinan Mab,
Jing Sun*b and
Zhibo Li*ab
aSchool of Polymer Science and Engineering, Qingdao University of Science and Technology, Qingdao 266042, China. E-mail: jingsun@qust.edu.cn; zbli@qust.edu.cn
bInstitute of Chemistry, Chinese Academy of Sciences, Beijing 100190, China
First published on 15th July 2016
Abstract
A series of poly(L-glutamate) bearing Y-shaped oligo(ethylene glycol)x (OEGx) side-chains (PPLG-g-EGx, x = 2, 3 and 4) were synthesized via a combination of ring opening polymerization (ROP) of γ-propargyl-L-glutamate N-carboxyanhydride (PLG-NCA) with thiol–yne photoaddition. The solubility of the polypeptides in water was firstly investigated. PPLG-g-EG3 and PPLG-g-EG4 were soluble in water and displayed fully reversible thermal-responsive behaviors. Additionally, the polypeptides showed redox-responsive properties along with the conformation associated water solubility due to the presence of thioether groups in the side-chains. In particular, the clouding points (CPs) of the polypeptides are highly tunable depending on the degree of polymerization (DP), the side-chain length, the polymer/salt concentration, and the degree of oxidation/reduction. The key feature of our design is the simultaneous incorporation of thermal-responsive OEG units and redox-responsive thioether linkages by one-step thiol–yne photoaddition, which allows for precisely tuning the phase transition temperature by both stimuli. This synthetic approach offers a facile and efficient way to prepare polypeptide-based biomaterials with highly tunable properties.
Introduction
Motivated by adaptive and responsive biological systems in nature, stimuli-responsive polymers have been extensively explored for many applications such as smart drug delivery systems, biosensors, and smart surfaces, etc.1–3 Particularly, polypeptide-based materials offer many advantageous properties for their excellent biocompatibility, biodegradability and unique assembling properties.4–6 The diversity of functional groups in the polypeptide side-chains is vast. The inherent ordered conformations (e.g., α-helices and β-sheets) of the polypeptides can respond to a variety of external stimuli and further trigger property changes.7–9 Considerable efforts have been made to develop efficient methods to prepare stimuli-responsive polypeptide-based smart materials.10,11Post-polymerization modification has emerged as a versatile and efficient strategy for the functionalization of polymers. The combination of living/controlled ROP of synthetic functional NCA monomers with orthogonal conjugation reactions, i.e., “Click” chemistry, greatly facilitates the preparation of stimuli-responsive polypeptides. Several NCA monomers with clickable substitutes have been prepared for subsequent modifications.12–18 In particular, alkynyl/vinyl groups modified polypeptides allow for the post-modification using thiol–yne/–ene photochemistry, respectively.19 Both reactions proceed rapidly and efficiently under ambient conditions without using metal catalysts, which offer significant advantages in biomedical applications.20,21 More importantly, the resultant thioether groups show redox-responsive behavior under mild conditions that often trigger polarity/conformation transitions of polypeptides. Deming and coworkers reported that the complete oxidation of poly(glyco-L-cysteine) and the partial oxidation of poly(L-methionine) can switch the secondary conformation from α-helix to random coil, and the partially oxidized poly(L-methionine) can be reduced back to the parent polymer.9,22 They also found that methionine-containing peptides can be alkylated by alkyl halides or epoxides into water-soluble disordered coils in high yields.23,24 Some of the dealkylated reactions were fully reversible. Our group previously synthesized a family of thermal-responsive OEGylated poly(L-cysteine)s via thiol–ene Michael addition.25 We further obtained stimuli-responsive self-assemblies that show oxidation-induced polarity/conformation changing.26
Combining the advantages of click chemistry with redox-responsive properties of the resulting thioether groups, we prepared a new class of OEGylated thioether bond-containing polypeptides derived from poly(L-glutamate)s (Scheme 1). The key feature of our design is the simultaneous incorporation of thermal-responsive OEG units and redox-responsive thioether bonds by thiol–yne photoaddition. The thiol–yne photoaddition allows for Y-shaped molecular architecture with two thermal-responsive OEG units on each side-chain. The redox-responsive properties of thioethers further provide an additional stimulus.
 | ||
Scheme 1 The synthetic route to PPLG-g-EGx by combination of ROP of PLG-NCA and thiol–yne photochemistry. Reagents and conditions: (i) HMDS, THF/DMF 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 v/v, rt, 48 h. (ii) DMPA, DMF, hν, 3 h. :1 v/v, rt, 48 h. (ii) DMPA, DMF, hν, 3 h. | ||
Both thermal-/redox-responsive properties allow access for highly tunable phase transition temperatures. This combination makes the obtained polypeptides promising candidates for multi-stimuli responsive polymeric materials.
Experimental section
Materials
Hexane, tetrahydrofuran (THF), and dichloromethane (DCM) were deoxygenated and dried by purging with nitrogen and passaging through activated alumina columns prior to use. Ethyl acetate (EtOAc) was freshly distilled from CaH2 prior to use. Anhydrous N,N-dimethylformamide (DMF) was purchased from Alfa Aesar and used under an inert and dry atmosphere. Deionized water (18 MΩ cm) was obtained from a Millipore Milli-Q purification unit. L-Glutamic acid was purchased from GL Biochem (Shanghai) Ltd. Hexamethyldisilazane (HMDS) was purchased from Sigma-Aldrich. 2,2-Dimethoxy-2-phenylacetophenone (DMPA) was purchased from Aladdin reagent. All other chemicals were purchased from commercial suppliers and used without further purification unless otherwise noted.Characterization
1H NMR spectra and 13C NMR spectra were recorded on Bruker AV400 FT-NMR spectrometer. All infrared spectroscopy measurements were performed using a Nicolet Avatar 330 FT-IR spectrometer. The solid samples was milled with potassium bromide (Aldrich) at mass ratio of 1:100, and pressed into disk before IR measurements. Tandem gel permeation chromatography/laser light scattering (GPC/LLS) was performed at 50 °C using an SSI pump connected to Wyatt Optilab DSP and Wyatt DAWN EOS light scattering detectors with 0.02 M LiBr in DMF as eluent at flow rate of 1.0 mL min−1. All samples for GPC/LLS were prepared at concentrations of 5–10 mg mL−1. The clouding points (CPs) were measured by monitoring the transmittance of a 500 nm light beam through a quartz sample cell at a concentration of 2 mg mL−1 on a Shimadzu UV-vis spectrometer. The solution was heated and cooled at a rate of 0.5 °C min−1. The CP was defined as the temperature corresponding to 50% transmittance of aqueous solution during the heating process. Circular dichroism (CD) spectra were recorded on a JASCO J-815 CD Spectropolarimeter. The solution was placed into a quartz cell with a path length of 1.0 mm with temperature controlled by a water bath, and the concentration of samples was 0.2 mg mL−1. Ellipticity ([θ] in deg cm2 dmol−1) = (millidegrees × mean residue weight)/(pathlength in millimetres × concentration of polypeptide in mg mL−1). The α-helix contents of the polypeptides were calculated using the following equation: % α-helix = (−[θ222] + 3000)/39000.27 The UV irradiation experiments were carried out using a mercury vapor lamp (254–577 nm, λmax = 365 nm, 500 W) as the UV-light source.
Synthesis of γ-propargyl-L-glutamate N-carboxyanhydride (PLG-NCA)
γ-Propargyl-L-glutamate (PLG) was prepared by direct coupling of propargyl alcohol and L-glutamic acid via sulfuric acid catalyzed esterification.28 PLG was purified according to previous work.29 After purification, the PLG monomer (5.0 g, 27 mmol) was converted into the corresponding N-carboxyanhydride (NCA) using triphosgene (2.9 g, 9.8 mmol) in THF (100 mL) at 50 °C under nitrogen. The solvent was removed under reduced pressure to yield slightly brown oil, which was subsequently re-dissolved in ethyl acetate and washed with ice-cold water and a 0.5% NaHCO3 ice-cold aqueous solution. The organic phase was then dried over anhydrous MgSO4 and evaporated to give PLG-NCA as light yellow oil with 40% yield.Synthesis of poly(γ-propargyl-L-glutamate) (PPLG)
PPLG was obtained by ROP of PLG-NCA using hexamethyldisilazane (HMDS) as the initiator (Scheme 1). All polymerizations were performed in a high-purity N2-filled glovebox. Typically, a solution of HMDS (0.1 mol L−1 in DMF) was added to a solution of PLG-NCA (50 mg mL−1 in THF/DMF 1:1 v/v). The reaction was stirred at room temperature, and polymerization progress was monitored by FTIR. Sample solutions were precipitated into diethyl ether. Solids were collected by centrifugation, which was further purified by dissolution in CH2Cl2 and precipitation from diethyl ether. The products were dried under reduced pressure to yield white sticky solid.
Synthesis of methoxy oligo(ethylene glycol) thiol modified PPLG by thiol–yne photochemistry (PPLG-g-EGx)
Methoxy oligo(ethylene glycol) thiol (OEGx-SH, x = 2, 3, 4) were synthesized using oligo(ethylene glycol) monomethyl ethers according to the literature procedures.30 A typical synthetic procedure is given for PPLG130-g-EG3 (the subscript 130 represents the average degree of polymerization of PPLG). PPLG130 (0.20 g, 1.20 mmol of alkynes), OEG3-SH (2.17 g, 12 mmol of thiols) and 2,2-dimethoxy-2-phenylacetophenone (DMPA, 31 mg, 0.12 mmol) were mixed in 4 mL DMF in a 20 mL vial under stirring. The reaction mixture was purged with N2 for 20 minutes before irradiated with UV light. The click reaction was performed at room temperature for 3 hours. The solution mixture was transferred to 3500 MWCO dialysis tubing and dialyzed against Millipore water for 3 days with water changing twice per day. Dialyzed polypeptides were lyophilized to give the product as a light yellow solid with 83% yield.General procedure for oxidation of PPLG-g-EGx to sulfoxides (PPLG-g-OEGx)
The oxidization experiments followed modified procedures as previously reported.9,22 A typical synthetic procedure is given for PPLG130-g-OEG3. PPLG130-g-EG3 (20 mg, 0.038 mmol of thioether groups) was dissolved in a mixed aqueous solution of 30% H2O2 (33 μL), AcOH (10 μL) and DI water (957 μL) and heated to 38 °C for 16 hours under stirring. Excess peroxide was quenched by 1 M sodium thiosulfate in water (0.33 mL). The solution mixture was transferred to a 3500 MWCO dialysis bag, and dialyzed against Millipore water for 3 days with water changing twice per day. Dialyzed polymers were lyophilized to yield partially oxidized products, PPLG130-g-OEG3 (93% yield).General procedure for oxidation of PPLG-g-EGx to sulfones (PPLG-g-O2EGx)
A typical synthetic procedure is given for PPLG130-g-O2EG3, PPLG130-g-EG3 (20 mg, 0.038 mmol of thioether groups) was dissolved in a mixed aqueous solution of 30% H2O2 (38 μL, 0.38 mmol) and AcOH (1 mL). The mixture was stirred at room temperature for 16 hours (about 20 °C). Excess peroxide was quenched by 1 M sodium thiosulfate in water (0.38 mL). The solution mixture was transferred to 3500 MWCO dialysis tubing and dialyzed against Millipore water for 3 days with water changing twice per day. Dialyzed polypeptides were lyophilized to yield completely oxidized products, PPLG130-g-O2EG3 (90% yield).General procedure for reduction of PPLG-g-OEGx
The reduction experiments followed reported procedures.7 PPLG-g-OEGx (x = 3 and 4) at 10 mg mL−1 was dissolved in DI water containing thioglycolic acid (10 eq. with respect of sulfoxide groups). The mixture was stirred at room temperature overnight and then diluted with equal amount water, followed by transferring to 3500 MWCO dialysis tubing. The solution mixture was dialyzed against Millipore water for 3 days with water changes twice per day. Dialyzed polypeptides were lyophilized to yield white solids.Results and discussion
γ-Propargyl-L-glutamate (PLG) and its corresponding NCA monomer were synthesized by modified procedures as reported.28,31 The obtained PLG-NCA was then polymerized in a mixture of THF/DMF (1:1 v/v) using HMDS as the initiator (Scheme 1).32 The chemical structures of the synthesized NCA and the corresponding homopolypeptides were confirmed by 1H NMR spectroscopy. All peaks were well assigned (Fig. 1a and b), indicating that the polymers were well prepared. The ratio of NCA to HMDS was varied to obtain polypeptides with different degree of polymerization (DP), summarized in Table 1.
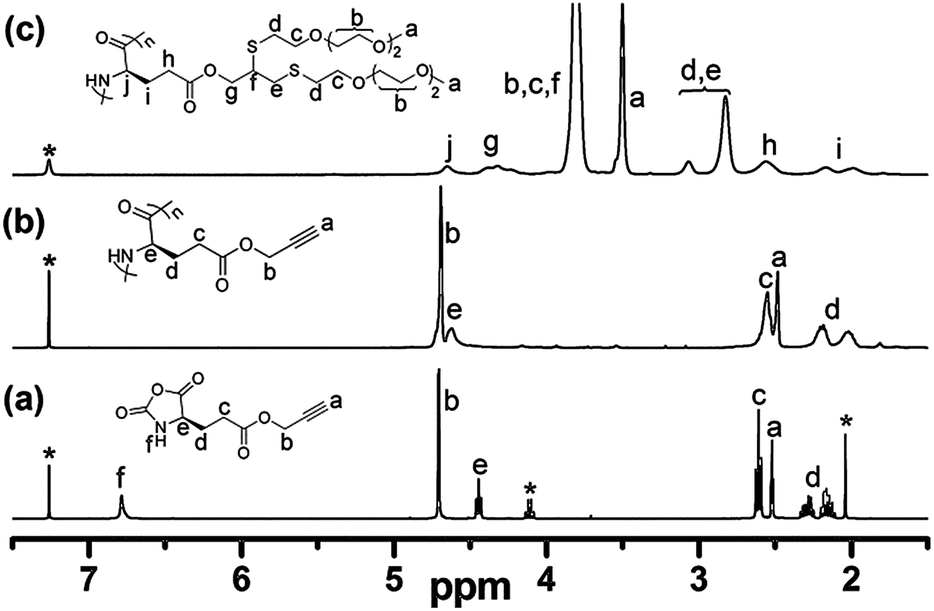 | ||
| Fig. 1 1H NMR spectra of (a) PLG-NCA in CDCl3 (* indicates CDCl3 and ethyl acetate residues); (b) PPLG and (c) PPLG-g-EG3 in CDCl3/CF3CO2D (v/v = 85:15). | ||
Three samples were synthesized, denoted as PPLG35, PPLG65 and PPLG130, where the subscript represented the DP of PPLG. GPC traces showed relatively narrow molecular weight distributions with dispersity (Ð) < 1.2 (Fig. S1a†), indicative of the well-controlled ROP of NCAs.
To systematically study the relationship of the OEG side-chain length on physical properties of poly(L-glutamate)s, we synthesized three types of thiol-terminated oligo(ethylene glycol) monomethyl ethers (OEGx-SH) with x ranging from 2 to 4. Their chemical structures were confirmed by 1H NMR spectrometry (see ESI†).30 OEGx-SH was then conjugated to PPLG via thiol–yne coupling reaction using DMPA as the photoinitiator in DMF. The reaction solution was purged with N2 to remove oxygen and irradiated with UV light at room temperature for 3 hours to yield the corresponding OEGx grafted homopolypeptides, PPLG-g-EGx.33 The chemical structure of PPLG-g-EGx was confirmed by 1H NMR spectroscopy. Fig. 1c shows a typical 1H NMR spectrum of PPLG-g-EG3. The protons of propargyl group at 4.66–4.76 and 2.46–2.53 ppm completely disappear, indicative of the quantitative conversion of alkynyl groups. The appearance of new peaks at 2.73–3.16, 3.50, and 3.80 ppm correspond to the protons of OEG moiety, further confirming the successful addition. The GPC traces of PPLG-g-EGx (Fig. 2a and S1†) maintain unimodal distribution with an apparent shift to high molecular region, indicative of negligible side reactions. FTIR spectra show that a characteristic –C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C– stretching band at 2130 cm−1 completely disappear (Fig. 2b and S2†), confirming the complete conversion of alkynyl groups. The obtained PPLG-g-EGx samples show good solubility in common organic solvents, but behave different in aqueous solution, summarized in Table S1.† PPLG-g-EG2 with the shortest OEG side-chain length is insoluble in water at ambient temperature; however, PPLG-g-EG3 and PPLG-g-EG4 are readily soluble under similar conditions. This is possibly due to the increased hydrophilicity of the polymers with longer OEG side-chains.34,35 We therefore focus on PPLG-g-EG3 and PPLG-g-EG4.
C– stretching band at 2130 cm−1 completely disappear (Fig. 2b and S2†), confirming the complete conversion of alkynyl groups. The obtained PPLG-g-EGx samples show good solubility in common organic solvents, but behave different in aqueous solution, summarized in Table S1.† PPLG-g-EG2 with the shortest OEG side-chain length is insoluble in water at ambient temperature; however, PPLG-g-EG3 and PPLG-g-EG4 are readily soluble under similar conditions. This is possibly due to the increased hydrophilicity of the polymers with longer OEG side-chains.34,35 We therefore focus on PPLG-g-EG3 and PPLG-g-EG4.
It is known that the secondary structure of polypeptides can be affected by both the main chain length and the side chain structures.36 We thus investigated the conformation of the samples using FTIR and circular dichroism (CD) spectroscopy. All of PPLG samples exhibit strong absorption bands centered at 1653 (amide I) and 1550 cm−1 (amide II), indicative of predominant helical conformation (Fig. S2a†). In addition, a broad band in the range of 1630–1660 cm−1 and a weak band at 1516 cm−1 were observed, indicative of the coexistence of β-sheet or random coil.37 As the PPLG chain length increases from 35 to 130, the absorbance at 1653 and 1550 cm−1 increases while decreasing at 1516 cm−1 in all cases, suggesting the enhanced helical content. This is consistent to previously reported results of OEGylated poly-L-glutamate (poly-L-EGxGlu).38 Interestingly, all PPLG-g-EGx samples show sharp absorption bands at 1653 (amide I) and 1550 cm−1 (amide II) and trivial absorption at 1516 cm−1 (Fig. 2b and S2†). This demonstrates that the introducing of OEGx side-chains is beneficial for stabilization of α-helical structures.29 Note that the conformation shows less dependence on OEG side-chain length. The secondary structures of PPLG-g-EG3 and PPLG-g-EG4 were also characterized by CD spectra, shown in Fig. 2c and S3.† We calculated the α-helical contents based on the CD analysis, and the results are given in Table S2.† CD measurement reveals that both PPLG-g-EG3 and PPLG-g-EG4 adopt α-helix conformation in water with 100% helicity at 20 °C. The helicity is nearly independent on OEG side-chain length and PPLG main-chain length in the case of PPLG-g-EG3 and PPLG-g-EG4, which is consistent with the FTIR results. These results are different from that of poly-L-EGxGlu, which show the increased helical content with increasing OEG side-chain length.29 This is possibly due to the increased OEG moiety in the Y-shape structure of PPLG-g-EGx that can stabilize the helicity of polypeptide backbones.
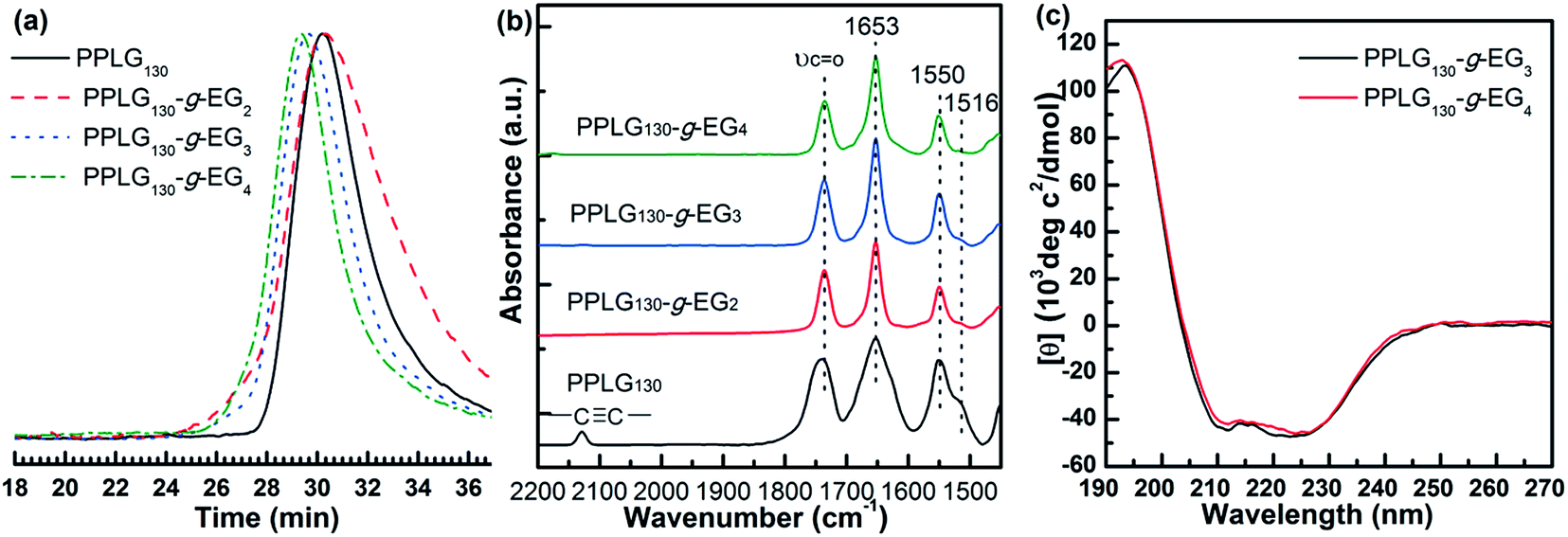 | ||
| Fig. 2 (a) GPC chromatographs and (b) FTIR spectra of PPLG130, PPLG130-g-EG2, PPLG130-g-EG3, and PPLG130-g-EG4; (c) CD spectra of PPLG130-g-EG3 and PPLG130-g-EG4 aqueous solutions (0.2 mg mL−1) at 20 °C. | ||
We then examined the effects of temperature on solution properties of samples PPLG-g-EG3 and PPLG-g-EG4 in water. In both cases, fast transitions from clear to cloudy upon heating and cloudy to clear upon cooling were shown, indicative of reversible LCST behaviors. Turbidimetry was applied to determine the corresponding cloud points (CPs) (Fig. 3a and S4†). Fig. 3a shows the transmittance versus temperature plots of PPLG130-g-EG3 and PPLG130-g-EG4 aqueous solutions (2 mg mL−1). In the case of PPLG130-g-EG3, the transmittance decreases from 100 to 1% when temperature increases from 5 to 45 °C and recovers to 100% upon cooling. If we define the temperature with 50% transmittance as CP, the CP of PPLG130-g-EG3 is about 25 °C for the heating process. Similarly, PPLG130-g-EG4 shows a sharp phase transition with the corresponding CP of 42 °C and complete recovery of transmittance upon cooling. Note that the CP determined from the cooling ramp is about 2 °C lower than that from the heating ramp in both cases. This is probably because rehydration of OEG units requires slight overcooling to overcome the energy barriers.39 We further studied the repeatability of phase transitions, shown in Fig. 3b. A full recovery of transmittance in PPLG130-g-EG3 was achieved after 6 heating–cooling cycles between 5 and 35 °C. Similarly, the phase transition of PPLG130-g-EG4 could be repeated multiple times between 30 and 55 °C (Fig. S4c†).
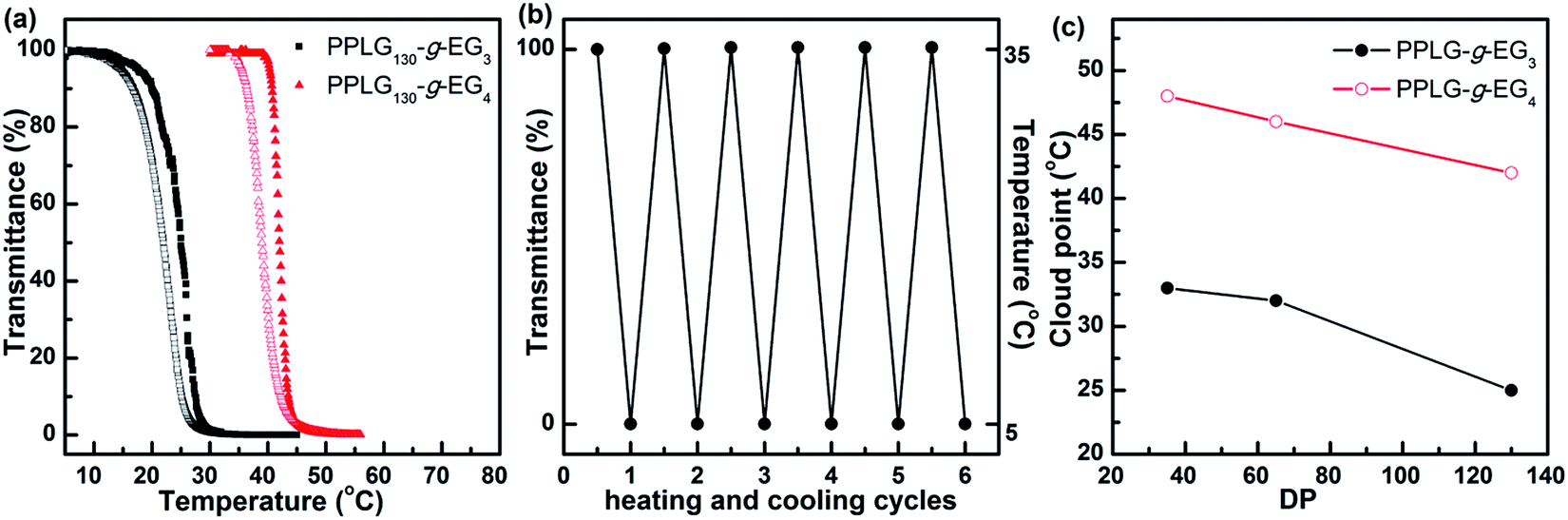 | ||
| Fig. 3 (a) Plots of transmittance as a function of temperature for aqueous solutions of PPLG130-g-EG3 and PPLG130-g-EG4. Solid symbols: heating ramp; open symbols: cooling ramp; 0.5 °C min−1. (b) Optical transmittance of PPLG130-g-EG3 solution versus 6 heating–cooling cycles between 5 and 35 °C; 2 °C min−1. (c) The cloud point dependence on DP for PPLGn-g-EG3 and PPLGn-g-EG4. All sample concentrations were 2 mg mL−1. | ||
Fig. 3c shows the CP dependence on DP for PPLGn-g-EG3 and PPLGn-g-EG4. It is revealed that PPLGn-g-EG3 shows a lower CP than that of PPLGn-g-EG4 at the same DP. It is not surprised as the increase of OEG length in the side-chains can increase the hydrophilicity of the polymer, resulting in the increase of CP. With the same OEG side-chain length, the CP decreases with increasing the DP of PPLG backbones. In the case of PPLGn-g-EG3, the CP decreases from 33 to 25 °C as the DP increases from 35 to 130. Similarly, the CP of PPLGn-g-EG4 decreases from 48 to 42 °C with increasing DP. The effects of polymer/salt concentration on the CP were also investigated. As the polymer concentration increases from 0.5 to 2 mg mL−1, the CP of PPLG65-g-EG3 decreases rapidly, accompanied by the reduced transition process (Fig. S5†). This is likely due to the enhanced intermolecular association in concentrated solution.31 When the polymer concentration further increased, the CP reaches a plateau. NaCl was added to investigate the effect of the salt concentration on CPs. As the NaCl concentration increases from 0 to 100 mM, the CP decreases slightly from 32 to 31.4 °C, which is possibly due to the partial dehydration of polymer chains induced by addition of NaCl (Fig. S6†).40 Hence, it is seen that the CP can be finely tuned by the OEGx side-chain lengths, the DP of PPLG backbone and the polymer/salt concentration for different requirements of biomedical applications.
The stability of the secondary structure of polypeptides on temperature was then investigated by temperature-varied CD measurements (Fig. 4 and S7–S9†). When the samples PPLGn-g-EG3 and PPLGn-g-EG4 were heated above the CPs, the CD signals significantly decreased (Fig. 4a and S7a–S9a†). The helicities declined accordingly, shown in Table S2.† This is most likely due to the aggregation when the temperature is above the CP.25 Their conformations are recovered during the cooling ramp (Fig. 4b and S7b–S9b†). The result is consistent with turbidity measurements.
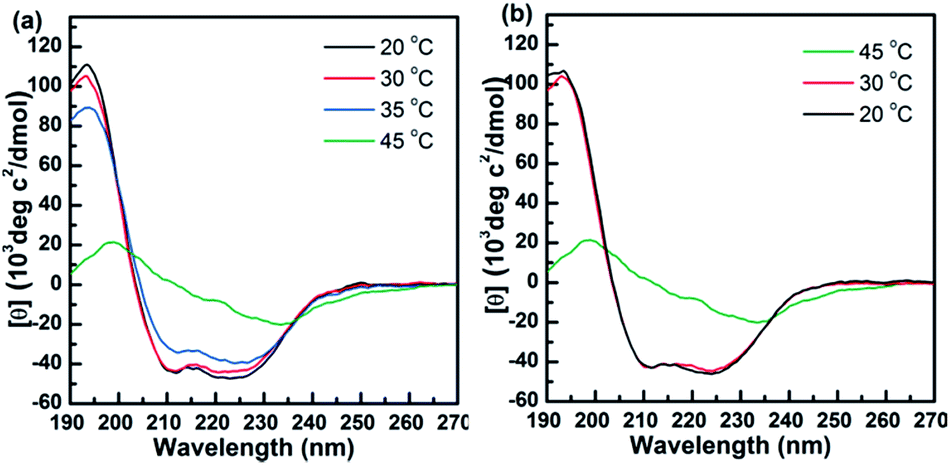 | ||
| Fig. 4 CD spectra of PPLG130-g-EG3 as a function of temperature (a) heating scan; (b) cooling scan. | ||
It is known that thioethers can undergo selectively oxidation into either sulfoxide or sulfone groups,9,25 while the partially oxidized sulfoxide polymers can be reversed to thioethers via enzymatic or selective chemical reduction (Scheme 2).7,22 We therefore performed different levels of chemical oxidation to achieve partially oxidized sulfoxide polymers (PPLG-g-OEGx) and fully oxidized sulfone polymers (PPLG-g-O2EGx). The corresponding structures were confirmed by 1H NMR and FTIR spectra. Fig. 5a shows the 1H NMR spectrum of PPLG-g-EG3, in which the broad peaks between 2.73 and 3.16 ppm are attributed to methylene groups (d and e) in β position to sulfur atoms (–CH2SCH(CHCH2)CH2SCH2–). As oxidized to sulfoxides (PPLG-g-OEG3), it is clearly observed that the peaks at 2.73–3.16 ppm disappear and the peaks at 3.11–3.60 ppm appear (Fig. 5b) that can presumably be ascribed to methylene groups in the β position of sulfoxides (–CH2S(O)CH(CHCH2)CH2S(O)CH2–). For PPLG-g-O2EG3, a higher-frequency shift of methylene groups (d and e in Fig. 5c) is observed, which can be assigned to more polar sulfone groups. FTIR spectra show increased νS![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O band at 1030 cm−1 of PPLG-g-OEG3 (Fig. 6a). In the case of PPLG-g-O2EG3, two types of new FTIR bands at 1291 and 1321 cm−1 appear, likely associated with the symmetric and asymmetric stretching of –SO2– groups. All the data indicate the successful oxidation to different levels.
O band at 1030 cm−1 of PPLG-g-OEG3 (Fig. 6a). In the case of PPLG-g-O2EG3, two types of new FTIR bands at 1291 and 1321 cm−1 appear, likely associated with the symmetric and asymmetric stretching of –SO2– groups. All the data indicate the successful oxidation to different levels.
 | ||
| Scheme 2 Structures and redox properties of PPLG-g-EGx homopolypeptides. | ||
 | ||
| Fig. 5 1H NMR spectra of (a) PPLG-g-EG3, (b) PPLG-g-OEG3, (c) PPLG-g-O2EG3 and (d) PPLG-g-*EG3 in CDCl3/CF3CO2D (v/v = 85:15). | ||
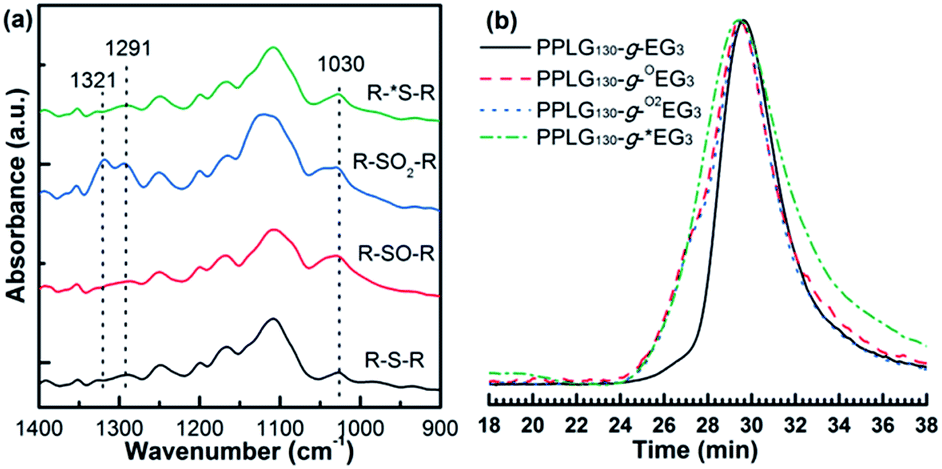 | ||
| Fig. 6 (a) FTIR spectra and (b) GPC chromatographs of PPLG130-g-EG3, PPLG130-g-OEG3, PPLG130-g-O2EG3 and PPLG130-g-*EG3. | ||
To investigate the reduction of sulfoxides, we initially incubated PPLG-g-OEGx with MSR enzymes (methionine sulfoxide reductase A and methionine sulfoxide reductase B) and DTT according to reported procedures.22 Unfortunately, we did not observe considerable reduction from PPLG-g-OEGx to PPLG-g-EGx. We assume that the synthetic polypeptide is not a good substrate for MSR enzymes. This may be ascribed into two possible reasons. One is that the specificity and selectivity of enzymes excludes the other different structures of substrates from natural methionine sulfoxides. The other possible reason is that the Y-shaped side-chain structure of PPLGn-g-EGx sterically limits the reduction efficiency of the sulfoxide groups. Beyond MSR enzymes, we also used thioglycolic acid to explore the reduction reaction of PPLG-g-OEGx. The structures of the reducing product, denoted as PPLGn-g-*EGx, were confirmed by 1H NMR and FTIR spectra (Fig. 5d, 6a and spectral data in ESI†). Fig. 5d shows a typical 1H NMR spectrum of PPLG130-g-*EG3. Two multiple peaks at 2.73–3.16 ppm and 3.11–3.60 ppm are clearly observed, indicative of the coexistence of sulfoxides and thioethers. We therefore increased the amount of reducing reagent (10 eq. with respect to sulfoxide groups). The degree of oxidation/reduction was quantitatively calculated based on the integrals of methylene peaks at 2.73–3.16 ppm and 3.11–3.60 ppm. FTIR data exhibit a decrease of infrared absorption band at 1030 cm−1 (νSO band, Fig. 6a) after reduction, consistent with the 1H NMR analysis. GPC traces demonstrated that the polypeptide backbones and ester groups on the side-chains remain intact under the oxidation or reduction reaction condition (Fig. 6b and S11†).
It is generally accepted that the thermal-responsive properties of the polymer in water is mainly governed by hydrophilic/hydrophobic balance of polymer subunits.7,9 As the thioether groups are oxidized into sulfoxide or sulfone groups, the molecular polarity and hydrophilicity of the system can be largely increased. We therefore investigated the influence of redox properties on solubility and CPs of PPLG130-g-EGx. PPLG130-g-EG2, insoluble in water at room temperature, shows reversible LCST behavior with the CP of 32 °C after partially oxidation (Fig. S15a†). The degree of oxidation was calculated to be 19% by 1H NMR. Fully conversion to PPLG130-g-OEG2 and PPLG130-g-O2EG2 remains soluble in water at elevated temperature up to 85 °C without showing thermal-responsive properties. This is possibly due to the increased hydrophilicity of polypeptides upon fully oxidation into sulfoxides or sulfones. Fig. 7a shows plots of transmittance versus temperature for PPLG130-g-EG3 at different oxidizing degree. As the OEG side-chain length increased to 3 and 4, the polypeptides are completely soluble in water without showing observable thermal-responsive behaviors up to water boiling point (Fig. S15b†). Similarly, the oxidation of PPLG65-g-EG3 and PPLG65-g-EG4 disrupt the thermal-responsive property of the polymers (Fig. S14†). Reduction of sulfoxide groups can recover thermal-responsive property as the redox reaction between sulfoxide and thioether groups is reversible. As shown in Fig. 7a, PPLG130-g-*EG3 after reduction by thioglycolic acid, denoted as PPLG130-g-*EG3 (1st), regains reversible LCST behavior. The CP of PPLG130-g-*EG3 (1st) is determined to be 38 °C, located between the CP of PPLG130-g-EG3 (25 °C) and the CP of PPLG130-g-OEG3 (above 80 °C), indicative of partial reduction of sulfoxide groups.7 We further calculated the degree of reduction to be 85% by 1H NMR. Similarly, reducing products PPLG130-g-*EG4, PPLG65-g-*EG3 and PPLG65-g-*EG4 also show reversible LCST behavior (Fig. S14 and S15†). To investigate the repeatability of redox process, we performed two oxidation/reduction cycles for PPLG130-g-EG3. It is observed that the reduction product in the second cycle, denoted as PPLG130-g-*EG3 (2nd), still show reversible LCST behavior with a CP at 52 °C, indicative of reversible redox properties. It is estimated that 75% of sulfoxide groups in PPLG130-g-*EG3 (2nd) are reduced to thioethers by 1H NMR.
 | ||
| Fig. 7 (a) Plots of transmittance as a function of temperature for of PPLG130-g-EG3 (2 mg mL−1) at different degrees of oxidation/reduction. Solid symbols: heating ramp; open symbols: cooling ramp; 0.5 °C min−1. * = partially oxidized samples after reduction using thioglycolic acid. (b) FTIR and (c) CD spectra of PPLG130-g-EG3, PPLG130-g-OEG3, PPLG130-g-O2EG3 and PPLG130-g-*EG3. | ||
It is known that the inherent secondary structures of polypeptides can largely influence the thermal-responsiveness.26,29 We therefore investigated the effects of secondary structures on redox properties of PPLGn-g-EGx using FTIR and CD spectroscopy. FTIR was first used to study the conformation in solid state at room temperature (Fig. 7b, S12b and S13b†). Generally, PPLG130-g-OEG3, PPLG130-g-O2EG3 and PPLG130-g-*EG3 show similar infrared absorption bands to that of PPLG130-g-EG3 with strong absorbance at 1653 cm−1(amide I) and 1550 cm−1 (amide II), indicative of predominant α-helical structures. We thus assume that the conformation remains during the redox process. The helicity of the polymers was determined by CD spectra. Fig. 7c shows that PPLG130-g-EG3, PPLG130-g-OEG3 and PPLG130-g-*EG3 exhibit similar CD traces with nearly 100% helicity, summarized in Table S3.† This is not surprised as the sulfoxide functionality is not sufficient to destabilize the ordered helical conformation.9,25 Further oxidation into PPLG130-g-O2EG3 results in the decrease of helicity from 100% to 69%. The possible reason is that conversion from thioethers to sulfones generally destabilizes the α-helices.38,41 The similar results were also observed in PPLG130-g-O2EG2, PPLG130-g-O2EG4, PPLG65-g-O2EG3 and PPLG65-g-O2EG4 (see ESI†). Note that the CD data are not quite in agreement with the FTIR results, possibly because FTIR measurements were performed in dry state. In the case of CD measurements, which were recorded in aqueous solution, the steric effects of solvated OEG side chains may dominate that result in destabilization of the helical conformation.25
Conclusion
In this article, we have reported the synthesis of poly(L-glutamate) with Y-shaped OEGx (x = 2–4) side-chains by combining the ROP of α-amino acid NCA with thiol–yne photochemistry. The obtained polypeptides of PPLG-g-EGx with x = 3 and 4 exhibit reversible thermal-responsive behavior. All the polypeptides display redox-responsive properties due to the presence of thioethers in the side-chains. The partially oxidized sulfoxide polymers can be reversibly reduced to thioethers; however, full oxidation to sulfone polymers is irreversible. The secondary structures and CPs were systematically investigated. We demonstrate that the CPs of the polypeptides are highly tunable depending on the degree of polymerization (DP), the length of OEG side-chains, the polymer concentration, the salt (e.g., NaCl) concentration and the degree of oxidation/reduction. This synthetic approach offers a facile and efficient way to prepare responsive polypeptide-based materials for potential biomedical applications.Acknowledgements
The authors appreciate financial support from the National Natural Science Foundation of China (20974112, 50821062).References
- S. Mura, J. Nicolas and P. Couvreur, Nat. Mater., 2013, 12, 991–1003 CrossRef CAS PubMed.
- M. A. Stuart, W. T. Huck, J. Genzer, M. Muller, C. Ober, M. Stamm, G. B. Sukhorukov, I. Szleifer, V. V. Tsukruk, M. Urban, F. Winnik, S. Zauscher, I. Luzinov and S. Minko, Nat. Mater., 2010, 9, 101–113 CrossRef PubMed.
- A. S. Hoffman, Adv. Drug Delivery Rev., 2013, 65, 10–16 CrossRef CAS PubMed.
- C. He, X. Zhuang, Z. Tang, H. Tian and X. Chen, Adv. Healthcare Mater., 2012, 1, 48–78 CrossRef CAS PubMed.
- H. Lu, J. Wang, Z. Song, L. Yin, Y. Zhang, H. Tang, C. Tu, Y. Lin and J. Cheng, Chem. Commun., 2014, 50, 139–155 RSC.
- Y. Shen, X. Fu, W. Fu and Z. Li, Chem. Soc. Rev., 2015, 44, 612–622 RSC.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2014, 136, 5547–5550 CrossRef CAS PubMed.
- J. Shen, C. Chen, W. Fu, L. Shi and Z. Li, Langmuir, 2013, 29, 6271–6278 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, J. Am. Chem. Soc., 2012, 134, 4112–4115 CrossRef CAS PubMed.
- S. Zhang and Z. Li, J. Polym. Sci., Part B: Polym. Phys., 2013, 51, 546–555 CrossRef CAS.
- T. J. Deming, Chem. Rev., 2016, 116, 786–808 CrossRef CAS PubMed.
- A. J. Rhodes and T. J. Deming, ACS Macro Lett., 2013, 2, 351–354 CrossRef CAS.
- Q. H. Xu, C. L. He, C. S. Xiao, S. J. Yu and X. S. Chen, Polym. Chem., 2015, 6, 1758–1767 RSC.
- J. R. Zhou, P. P. Chen, C. Deng, F. H. Meng, R. Cheng and Z. Y. Zhong, Macromolecules, 2013, 46, 6723–6730 CrossRef CAS.
- A. C. Engler, H. I. Lee and P. T. Hammond, Angew. Chem., Int. Ed., 2009, 48, 9334–9338 CrossRef CAS PubMed.
- J. Sun and H. Schlaad, Macromolecules, 2010, 43, 4445–4448 CrossRef CAS.
- Y. Xu, M. Zhu, M. Li, Y. Ling and H. Tang, Polym. Chem., 2016, 7, 1922–1930 RSC.
- E. G. Gharakhanian and T. J. Deming, J. Phys. Chem. B, 2016, 120, 6096–6101 CrossRef CAS PubMed.
- S. M. Brosnan and H. Schlaad, Polymer, 2014, 55, 5511–5516 CrossRef CAS.
- C. E. Hoyle, A. B. Lowe and C. N. Bowman, Chem. Soc. Rev., 2010, 39, 1355–1387 RSC.
- M. A. Quadir, M. Martin and P. T. Hammond, Chem. Mater., 2014, 26, 461–476 CrossRef CAS.
- A. R. Rodriguez, J. R. Kramer and T. J. Deming, Biomacromolecules, 2013, 14, 3610–3614 CrossRef CAS PubMed.
- E. G. Gharakhanian and T. J. Deming, Biomacromolecules, 2015, 16, 1802–1806 CrossRef CAS PubMed.
- J. R. Kramer and T. J. Deming, Chem. Commun., 2013, 49, 5144–5146 RSC.
- X. Fu, Y. Shen, W. Fu and Z. Li, Macromolecules, 2013, 46, 3753–3760 CrossRef CAS.
- X. Fu, Y. Ma, Y. Shen, W. Fu and Z. Li, Biomacromolecules, 2014, 15, 1055–1061 CrossRef CAS PubMed.
- J. A. Morrow, M. L. Segall, S. Lund-Katz, M. C. Phillips, M. Knapp, B. Rupp and K. H. Weisgraber, Biochemistry, 2000, 39, 11657–11666 CrossRef CAS PubMed.
- C. Xiao, C. Zhao, P. He, Z. Tang, X. Chen and X. Jing, Macromol. Rapid Commun., 2010, 31, 991–997 CrossRef CAS PubMed.
- C. Chen, Z. Wang and Z. Li, Biomacromolecules, 2011, 12, 2859–2863 CrossRef CAS PubMed.
- A. W. Snow and E. E. Foos, Synthesis, 2003, 509–512 CrossRef CAS.
- Y. L. Cheng, C. L. He, C. S. Xiao, J. X. Ding, X. L. Zhuang and X. S. Chen, Polym. Chem., 2011, 2, 2627–2634 RSC.
- H. Lu and J. Cheng, J. Am. Chem. Soc., 2007, 129, 14114–14115 CrossRef CAS PubMed.
- Y. Huang, Y. Zeng, J. Yang, Z. Zeng, F. Zhu and X. Chen, Chem. Commun., 2011, 47, 7509–7511 RSC.
- Z. B. Hu, T. Cai and C. L. Chi, Soft Matter, 2010, 6, 2115–2123 RSC.
- J. F. Lutz, Adv. Mater., 2011, 23, 2237–2243 CrossRef CAS.
- H. Y. Tang and D. H. Zhang, Polym. Chem., 2011, 2, 1542–1551 RSC.
- P. I. Haris and D. Chapman, Biopolymers, 1995, 37, 251–263 CrossRef CAS PubMed.
- S. Zhang, C. Chen and Z. Li, Chin. J. Polym. Sci., 2013, 31, 201–210 CrossRef CAS.
- J.-F. Lutz, K. Weichenhan, Ö. Akdemir and A. Hoth, Macromolecules, 2007, 40, 2503–2508 CrossRef CAS.
- J. F. Lutz, O. Akdemir and A. Hoth, J. Am. Chem. Soc., 2006, 128, 13046–13047 CrossRef CAS PubMed.
- J. X. Ding, C. S. Xiao, L. Zhao, Y. L. Cheng, L. L. Ma, Z. H. Tang, X. L. Zhuang and X. S. Chen, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 2665–2676 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: GPC, CD, UV-vis, FTIR and NMR spectra of the samples. See DOI: 10.1039/c6ra17427b |
| This journal is © The Royal Society of Chemistry 2016 |