
Visible light assisted electro–photo synergistic catalysis of heterostructured Pd–Ag NPs/graphene for methanol oxidation
Abstract
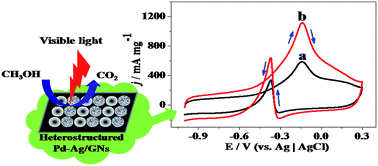
In this work, a facile method was used to synthesize the heterostructured bimetallic catalyst Pd–Ag/graphene (simplified as Pd–Ag/GNs). In order to achieve the heterostructured Pd–Ag nanoparticles (NPs), Ag2O was firstly produced on GNs from AgOH under UV irradiation, then the heterogeneous phase between the precursor Pd2+ and Ag2O contributes to the formation of the highly dispersed heterostructured Pd–Ag NPs with the average diameter of 5 nm on GNs after a synchronous reduction process. Electro-catalytic performances of Pd–Ag/GNs were investigated by cyclic voltammetry (CV), chronoamperometry, COad stripping voltammetry, and electrochemical impedance spectroscopy (EIS). It is shown that Pd–Ag/GNs has a much higher catalytic activity (595 mA mg−1) and stability for methanol oxidation reaction (MOR) and improved tolerance of CO compared with the counterpart Pd/GNs and the commercial 20% Pd/C catalyst (Pd/C-SA). And under visible light irradiation, the catalytic activity and stability of Pd–Ag/GNs are remarkably enhanced. Especially, its mass activity is 1128 mA mg−1 which is 1.9 times higher than that without light irradiation. The significantly improved performance benefits from the efficient electro–photo synergistic catalysis for MOR under visible light irradiation, which is in favor of harvesting sunlight to produce clean energy.
 Please wait while we load your content...
Please wait while we load your content...

