
Dynamic vulcanization of castor oil in a polylactide matrix for toughening
Tong-Hui Zhao,
Yan He,
Yi-Dong Li*,
Ming Wang and
Jian-Bing Zeng*
School of Chemistry and Chemical Engineering, Southwest University, Chongqing 400715, China. E-mail: dongdongdong555@sina.com; jbzeng@swu.edu.cn; Tel: +86-23-68254000
First published on 15th August 2016
Abstract
Dynamic vulcanization of biomass-derived castor oil in the presence of 4,4′-diphenylmethane diisocyanate (MDI) in a polylactide (PLA) matrix was performed with the aim of toughening PLA sustainably. Crosslinking of castor oil with MDI took place rapidly under dynamic vulcanization conditions, leading to the formation of a tough blend, which showed a phase-separated morphology with castor oil derived polyurethane (COP) as a dispersed phase and PLA as a continuous phase. The content of COP in the blends played an important role not only in the size of dispersed COP particles but also in the mechanical properties, rheological and crystallization behaviors of the formed PLA/COP blends. The size of dispersed COP, melt viscosity, and storage modulus of the blends increased significantly with COP content. The crystallization rate of PLA was enhanced by incorporation of COP due to the increased nucleation effect arising from interfacial nucleation of the phase-separated blends. The thermal stability of the blends was slightly reduced compared to neat PLA. The elongation at break of PLA was considerably increased by 45 times to 338%, compared to 7.5% of neat PLA, with the addition of only 5 wt% COP; meanwhile, the mechanical strength and modulus were largely retained. Cavitation, arising from dispersed phase debonding from the matrix, induced matrix plastic deformation was the toughening mechanism for the present PLA/COP blends.
1. Introduction
Biodegradable polymers, especially those derived from sustainable resources, have attracted increasing interest due to the increased resource crisis and environmental concerns associated with traditional petroleum-based and non-degradable polymers.1–3 Polylactide (PLA) is regarded as one of the most promising biobased and biodegradable polymers as it combines abundant advantages such as excellent biodegradability, biocompatibility, high mechanical strength, high melting temperature and easy processibility.4,5 However, PLA has some significant shortcomings, such as the inherent brittleness, slow crystallization rate and low heat distortion temperatures, which have largely restricted the wide application of PLA.6–8 Therefore, increasing efforts have been made to overcome the shortcomings of PLA.9With respect to the brittleness, many approaches, such as plasticization, copolymerization, polymer blending etc. have been explored to toughen PLA.9,10 Among those approaches, polymer blending with flexible or rubbery polymers provides a simple and cost-effective way of toughening PLA without changing the chemical structure and significant compromise with respect to its other excellent physical properties.10 The flexible polymers in the early studies were usually from petroleum-based resources, which would reduce the sustainability of the toughened PLA blends.11–13 In this sense, the flexible polymers used for toughening PLA in most of recent investigations have been transformed to those derived from renewable resources.14–19 It should be noted that most of the flexible polymers were immiscible with PLA, thus sufficient toughening efficiency is unavailable for the blends without compatibilization. Therefore, many strategies, including addition of copolymers, introduction of reactive small chemicals or polymers, importation of nanoparticles, interchange reactions, dynamic vulcanization and etc. have been employed in compatibilizing PLA-based blends, as summarized in our recent review article.20 Among those techniques, dynamic vulcanization represents a very efficient way not only in compatibilizing PLA-based blends but also in improving toughness of the resultant blends, as most supertough PLA-blends in recent literatures have been achieved by dynamic vulcanization.21–24
Dynamic vulcanization involves a process of selective vulcanization of a rubber phase during melt blending with a thermoplastic polymer, leading to a two-phase material in which the vulcanized rubber phase dispersed in the thermoplastic matrix.25 The coalescence of dispersed rubber domains is prevented by vulcanization, and thus the final morphology and physical properties of the resultant blends are fixed after dynamic vulcanization.20 It is well-known that the final properties of phase-separated polymer blends depend strongly on their morphologies, which are tunable in a wide range during dynamic vulcanization via various techniques depending on the specific blending systems. For example, the morphologies of a PLA ternary polymer blends with ethylene-butyl acrylate-glycidyl methacrylate (EBA-GMA) terpolymer and a zinc ionomer of ethylene methacrylic acid copolymer (EMAA-Zn) were tuned via selection of different dynamic vulcanization temperature and feed ratio of EBA-GMA/EMAA-Zn, which affected the reaction levels of both dynamic vulcanization and interfacial compatibilization;21,22 the morphologies of dynamic vulcanization system consisting of PLA and an unsaturated aliphatic polyester elastomer (UPE) were regulated by the content of initiator, which influenced gel fraction, particle size and shape of the vulcanized UPE phase;24 for dynamic vulcanization system consisting of PLA and natural rubber (NR) or epoxidized natural rubber (ENR), the morphologies could be well controlled by feed ratio and even cocontinuous morphology with supertoughness was obtained for some specific blending composition.23,26–30 Therefore, dynamic vulcanization is a very powerful way of tailoring morphology and physical properties of rubbery polymers toughened PLA blends. It is noted that vulcanization of rubbery polymers was usually proceeded via free radical initiation in most of PLA involved dynamic vulcanization systems.23,24,26–33 Few works21,22 focused on the vulcanization of rubbery polymers through other ways, for example, step polymerization, probably due to the slow vulcanization rate of most step polymerization systems.
Castor oil, a biomass derived triglyceride, containing about 90% of hydroxylated unsaturated fatty acid called ricinoleic acid, has been widely used in preparation of various sustainable polymers.34 It can be directly used as a toughening agent for PLA and the toughening efficiency could be improved if a poly(ricinoleic acid)–PLLA diblock copolymer was added as a compatibilizer, the blend containing 5 wt% castor oil and 5 wt% block copolymer showed elongation at break of ∼60%.14 However, castor oil would emigrate from PLA matrix to embrittle the toughened materials during service or storage. Some castor oil based copolymers, such as branched poly(castor oil)–poly(L-lactide) (PCO-g-PLLA) copolymers,35 poly(L-lactide)-b-poly(ricinoleic acid)-b-poly(L-lactide) (PLLA-b-PRic-b-PLLA) triblock copolymers,36 castor oil-based polyurethane prepolymer,37 and polyester-urethane networks consisting of castor oil and poly(ε-caprolactone),38 have been designed to toughen PLA. Although some of them showed good toughening efficiency, the cost performance of the obtained blends seems insufficient, due to the lengthy and complicated synthetic procedures of the copolymers. Therefore, a convenient and cost-effective way is still required to toughen PLA with biomass-derived castor oil derivatives for practical application.
Castor oil contains several hydroxyl groups, which is reactive towards isocyanate groups to form polyurethane.39 Although the reactivity of the secondary hydroxyl groups may not high compared to other primary hydroxyl groups, the processing temperature of PLA is high enough, for example 180 °C, at this temperature the reaction between secondary hydroxyl groups of castor oil and isocyanate groups may have a suitable reaction rate. Therefore, dynamic vulcanization of castor oil in the presence of a diisocyanate may be applicable in toughening PLA, which would then provide a facial and cost-effective way to toughen PLA with castor oil based products. The focus of this study is to evaluate the possibility of using dynamic vulcanization to toughen PLA with a diisocyanate vulcanized castor oil polyurethane (COP) as the toughening component. Therefore, the dynamic vulcanization of castor oil in PLA matrix with 4,4′-diphenylmethane diisocyanate as a crosslinking agent was performed first and then the morphology, thermal properties, rheological and crystallization behaviors, and mechanical properties of the resultant polymer blends were systematically investigated to disclose the insight in toughening modification of PLA with dynamically vulcanized castor oil polyurethane.
2. Experimental section
2.1. Materials
Castor oil with AR grade was obtained from Kelong Chemical Reagent Factory (Chengdu, China) and used as received. 4,4′-Diphenylmethane diisocyanate (MDI, 98%) was procured from Micxy Chemical Co., Ltd (Chengdu, China) and used without any purification. Polylactide (PLA 4032D) was purchased from NatureWorks LLC and dried at 80 °C oven for 8 h prior to use. Chloroform with AR grade was obtained from the Kelong Chemical Reagent Factory. All other chemicals with AR grade were used directly.2.2. Sample preparation
Dynamic vulcanization of castor oil with MDI in PLA matrix was performed in a Hapro torque rheometer (Harbin China) with two rollers within the mixing chamber. PLA and castor oil with predetermined amounts were first premixed in the rheometer at 180 °C and 50 rpm for 5 min to obtain uniform melts. Then dynamic vulcanization of castor oil occurred by addition of a certain amount of MDI at the same temperature and rotation rate. With the dynamic vulcanization occurred, the melt torque increased first and then levelled off (∼5 min), which was used to indicate the end of the dynamic vulcanization. The molar ratio of –NCO group of MDI to –OH group of castor oil was fixed at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Four samples with PLA weight fraction of 95%, 90%, 80% and 70% were prepared. The reaction of castor oil with MDI formed a polyurethane, which was named after castor oil based polyurethane (abbr. COP). For brevity, the four samples were abbreviated to PLA/COP-5, PLA/COP-10, PLA/COP-20 and PLA/COP-30, respectively. For property comparison, neat PLA was also treated with the same processing conditions. Standard tensile specimen (ASTM D638, type III) and Notched Izod impact (ASTM D256) specimen with 45° and 2 mm depth notch were prepared by injection molding (Xinshuo MiniJet, Shanghai, China) operated with a nozzle temperature of 180 °C and a molding temperature of 40 °C. The samples were dried in a vacuum oven at 80 °C for 8 h prior to injection molding.
:1. Four samples with PLA weight fraction of 95%, 90%, 80% and 70% were prepared. The reaction of castor oil with MDI formed a polyurethane, which was named after castor oil based polyurethane (abbr. COP). For brevity, the four samples were abbreviated to PLA/COP-5, PLA/COP-10, PLA/COP-20 and PLA/COP-30, respectively. For property comparison, neat PLA was also treated with the same processing conditions. Standard tensile specimen (ASTM D638, type III) and Notched Izod impact (ASTM D256) specimen with 45° and 2 mm depth notch were prepared by injection molding (Xinshuo MiniJet, Shanghai, China) operated with a nozzle temperature of 180 °C and a molding temperature of 40 °C. The samples were dried in a vacuum oven at 80 °C for 8 h prior to injection molding.
2.3. Characterizations
Fourier transform infrared (FT-IR) spectra of the samples under reflective mode were recorded on RF-5301PC spectrophotometer (Shimadzu, Japan) in a range of wavenumbers from 4000 to 400 cm−1 with the resolution and scanning time of 4 cm−1 and 32 times, respectively.The gel fraction of the sample was measured by solvent extraction. The sample with predetermined weight (w1, ∼2 g) was immersed in 100 mL chloroform at room temperature for a week to dissolve the soluble part. The undissolved part was isolated from the solution and washed with chloroform for three times. The weight (w2) was measured after drying at 80 °C for 12 h. Then the gel fraction was calculated by w2/w1 × 100%.
The soluble part was precipitated in excessive methanol and vacuum dried at 80 °C for 12 h. The proton nuclear magnetic resonance (1H NMR) spectra of neat PLA and the precipitated PLA were recorded on a Bruker AC-P 400 MHz spectrometer at room temperature with CDCl3 and tetramethylsilane (TMS) as a solvent and an internal chemical shift standard, respectively.
Rheological behaviors of neat PLA and PLA/COP blends with different compositions were measured on a TA DHR-1 rotational rheometer in dynamic frequency sweep mode from 0.1 to 100 rad s−1 at 180 °C with an oscillation strain of 1.0%.
Differential scanning calorimetry analysis was performed on a NETZSCH instruments DSC-214. The samples with ∼6 mg in aluminum DSC pans were first melted at 190 °C for 3 min to remove thermal history, then quenched to 0 °C at a cooling rate of 80 °C min−1, and finally heated up to 190 °C at a heating rate of 10 °C min−1 after maintaining at 0 °C for 3 min. All the processes were conducted under N2 atmosphere. The last heating curve was recorded for analysis.
Spherulitic morphologies of neat PLA and PLA/COP blends were studied with an Olympus BX51 polarized optical microscope (POM) equipped with an HT600 hot stage. Sample films were pressed between two microscopic cover glasses at 190 °C. The samples were first melted at 190 °C for 3 min to eliminate the thermal history and quenched to the temperature of 120 °C to isothermally crystallize. The final morphology was recorded for analysis.
Scanning electron microscopy (SEM) was used to observe the morphologies of the samples. The cryo-fractured surfaces of injection-molded specimens were observed with an XL-30s FEG SEM (Philips, Holland) at an accelerating voltage of 5 kV, and the tensile fractured surfaces were observed by a JSM-6510 SEM (JEOL, Japan) at an accelerating voltage of 5 kV. The surfaces were sputtered with a layer of gold prior to observation. Thermal stability of the samples under N2 atmosphere was performed on a TA TGA-Q600 from room temperature to 700 °C at a heating rate of 10 °C min−1.
Tensile properties of the samples were measured with a Sansi Universal Testing Machine (CMT6503) at a crosshead speed of 5 mm min−1 at room temperature. The notched Izod impact test was performed on a Sansi ZBC7000 (Shenzhen, China) impact tester at room temperature.
3. Results and discussion
3.1. Preparation, characterization and morphologies of PLA/COP blends
Dynamic vulcanization was used to prepare PLA/COP blends, in which the COP was in situ formed by polymerization of castor oil and MDI, as shown in Scheme 1. The dynamic vulcanization was performed in a torque rheometer. For comparison, neat PLA was also processed with the same condition. The melt torque versus processing time was recorded to monitor the dynamic vulcanization process, as shown in Fig. 1. To obtain well dispersed phase morphology of the blends, castor oil and PLA were first premixed at 180 °C for ∼5 min, and then predetermined amount of MDI was added into the mixing chamber to proceed dynamic vulcanization. It is obvious that before addition of MDI, the melt torque of the sample decreased with increasing castor oil content, due to the lubrication or plasticization effects of low viscosity castor oil; after addition of MDI, the melt torque increased firstly, indicative of vulcanization of castor oil with MDI, and then levelled off, indicating the completion of vulcanization. Both the increment in melt torque and the final melt torque of the sample increased with increasing COP content, which is attributed to the increased overall melt viscosity and reduced chain mobility resulted from the increase in the content of crosslinking part of the sample. By comparison, the melt torque of PLA decreased slightly with processing time, indicative of somewhat thermal degradation during high temperature shearing.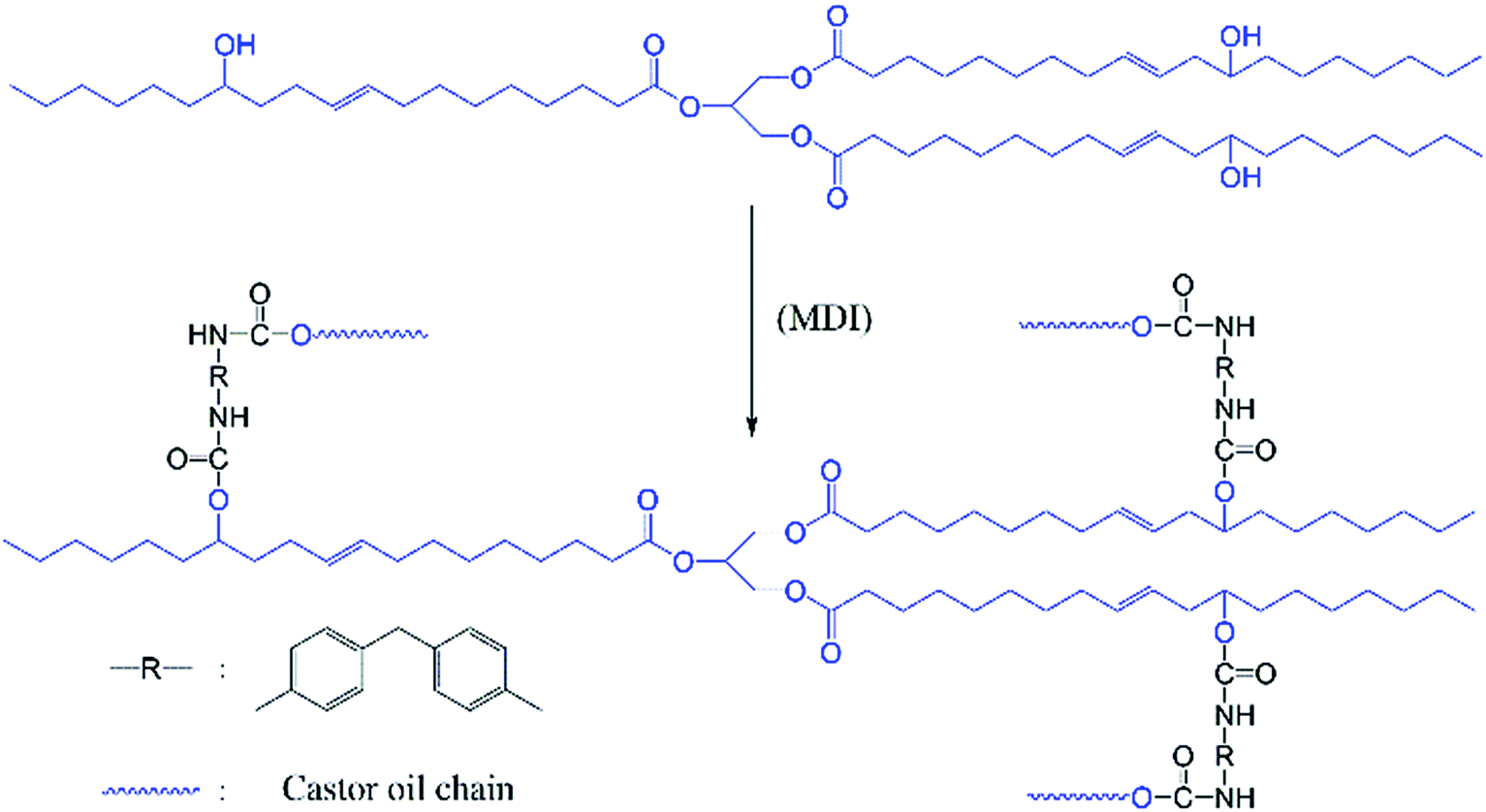 | ||
| Scheme 1 Reaction of castor oil with MDI to form castor oil based polyurethane during dynamic vulcanization. | ||
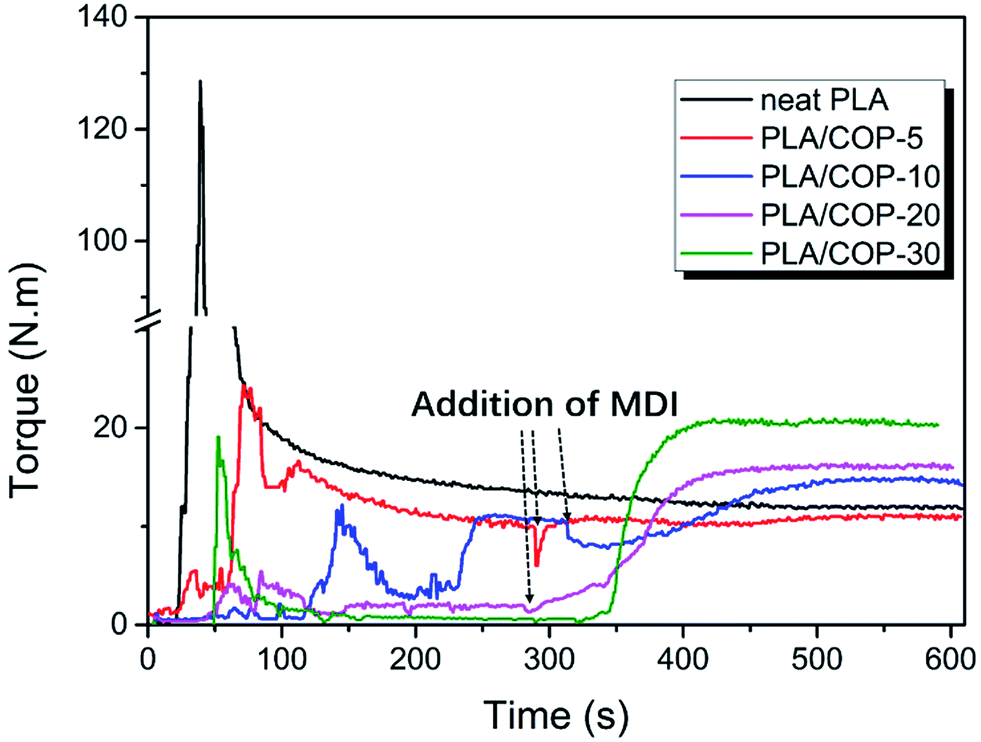 | ||
| Fig. 1 Melt torque versus time for processing neat PLLA and PLA/COP blends. | ||
Dynamic vulcanization of castor oil and MDI would form crosslinking polyurethane, which is insoluble in solvent. Therefore, we isolated the crosslinking part of the samples using chloroform as the solvent and measured the gel fraction of the samples. The values were 5.03 ± 0.05%, 9.04 ± 0.14%, 18.46 ± 0.12% and 28.36 ± 0.53% for PLA/COP-5, PLA/COP-10, PLA/COP-20 and PLA/COP-30, respectively. The gel fraction values were very close to the feed content of castor oil plus MDI, the smaller difference would be ascribed to inevitable loss of the COP during isolation process and some possible soluble branched products. The results indicate that castor oil and MDI reacted successfully to form cross-linked polyurethane.
In order to further confirm successful formation of polyurethane, FT-IR spectra of isolated crosslinking products, the as prepared blends and neat PLA were recorded, as shown in Fig. 2a. For the isolated COP, the characteristic absorption peaks of N–H, C–H, benzene ring and urethane linkage were observed at around of 3541, 2920–2854, 1596 and 1525 cm−1, respectively.3 Most of the characteristic absorptions could be observed in the spectra of the as prepared PLA/COP blends but were unavailable in that of neat PLA, indicating the formation of polyurethane in PLA matrix during dynamic vulcanization. In addition, no peak ascribing to the isocyanate group could be observed at around 2200 cm−1, indicating that MDI has been consumed completely.40,41 It is noted that PLA has terminal hydroxyl and carboxyl groups, which are also reactive towards isocyanate group.37 However, it is hard to find direct evidence for the reaction between PLA and isocyanate group from the FT-IR spectrum of the isolated COP. Therefore, we precipitated the soluble part of the PLA blends and ran NMR for the precipitates. The resonance signals of MDI and castor oil could be found from the 1H NMR spectrum if PLA reacted with MDI. Unfortunately, no resonance signal of both MDI and castor oil could be found from 1H NMR spectrum of precipitated PLA, as shown in Fig. 2b. Because no evidence for the reaction between isocyanate and PLA can be found, this reaction could be reasonably neglected compared to that between isocyanate and castor oil. The equivalent mole number of isocyanate to hydroxyl group of castor oil may cause complete consumption of isocyanate due to the relatively higher reactivity of low-molecular-weight castor oil compared to high-molecular-weight PLA. Consequently, no extra isocyanate was available for reaction with PLA, leading to undetectable reaction between isocyanate and PLA.
 | ||
| Fig. 2 (a) FT-IR spectra of neat PLA, isolated COP, and PLA/COP blends; (b) 1H NMR spectra of neat PLA and precipitated PLA. | ||
For binary polymer blends, the phase morphology plays an important role in the final physical properties. We observed the phase morphology of the PLA/COP blends by SEM. Fig. 3 shows the SEM images for the cryo-fractured surfaces of neat PLA and PLA/COP blends. Neat PLA showed smooth surface (Fig. 3a) while all blends showed rough surfaces with phase separation occurred (Fig. 3b–e). Both raised particles (red circle) and voids (red arrow) could be observed for the blends, which suggested that pulling out of dispersed COP phase occurred during cryo-fracturing, also indicating a low adhesion between dispersed COP phase and the continuous PLA matrix.
 | ||
| Fig. 3 SEM images for cryo-fractured surfaces of neat PLA (a and a′), PLA/COP-5 (b and b′), PLA/COP-10 (c and c′), PLA/COP-15 (d and d′), and PLA/COP-20 (e and e′) with magnification of 5000× (a–e) and 10000× (a′–e′). | ||
It is worth noting that the size of the dispersed COP phase depended strongly on its concentration, i.e., increasing apparently with COP content, which is very close to the morphologies of some other PLA based blends with rubbery polymers.17,19,42 To well observe the size of dispersed COP, the SEM images with higher magnification (10000×) of the PLA blends are given in Fig. 3b′–e′. For PLA/COP-5, the COP particles (marked with red circle) dispersed uniformly in PLA matrix. The size of most particles were at the scale of submicron and a few particles with size of 1–2 μm were also observed. When the content of COP increased to 10 wt%, the size of most of the observed COP particles were in the range of 1–3 μm. With further increasing COP content to 20 wt% and 30 wt%, large COP particles with a size of greater than 10 μm were observed. The increase in the size of COP phase with content was resulted from the coalescence of COP induced by the immiscibility between dispersed COP phase and continuous PLA matrix.
3.2. Rheological behavior of PLA/COP blends
During dynamic vulcanization process, the final melt torque of the blends increased with COP content, preliminarily indicating the increase in melt strength resulted from the formation of more content crosslinking parts in the blends. In order to study the effect of COP content on the rheological behaviors of PLA/COP blends, small amplitude oscillatory frequency sweeps were carried out at temperature of 180 °C to evaluate the effect of COP content on the complex viscosity, storage modulus, loss modulus, and loss tangent of the blends.The viscosity and the shear-shinning behavior of neat PLA would be changed by incorporation of vulcanized castor oil polyurethane. The variation of complex viscosity (|η*|) versus angular frequency (ω) for neat PLA and the PLA/COP blends with different compositions are shown in Fig. 4a. It is obvious that the complex viscosity at low frequency increased with COP content. Two types of angular frequency dependence of complex viscosity could be observed for the blends. For neat PLA and the blends with low COP content (below 20 wt%), an obvious Newtonian plateau could be observed at middle frequency range and an obvious reduction in complex viscosity occurred at high frequency range, indicative of a transition from the Newtonian plateau to power law regime at the inflection point. In the cases of the blends with high COP content (20 wt% and 30 wt%), the Newtonian plateau was hard to observe in the angular frequency range considered in this study as illustrated by the absence of frequency-independent viscosity, suggesting that the blends with high COP content followed more obviously the non-Newtonian flow behavior at the low frequency range. The shear-thinning behavior became stronger with increasing COP content of PLA/COP blends. The above phenomenon was related to the phase structure of PLA/COP blends through formation of cross-linked castor oil polyurethane particles in PLA matrix by dynamic vulcanization. Similar variation of complex viscosity versus frequency was also found in PLA blends with cross-linked poly(ethylene glycol)diacrylate (CPEGDA) formed by in situ crosslinking.43
 | ||
| Fig. 4 Variation of complex viscosity (a) storage modulus, G′ (b) and loss modulus, G′′ (c) and tanδ (d) versus angular frequency at 180 °C for neat PLA and PLA/COP blends with different composition. | ||
Except for complex viscosity, the storage modulus G′ and loss modulus G′′ were also sensitive to the composition of the PLA/COP blends. Fig. 4b and c shows the variation of G′ and G′′ versus angular frequency (ω). Both G′ and G′′ of all sample increased with increasing ω. PLA/COP blends showed higher G′ and G′′ than neat PLA in the full frequency range. With increasing COP content, both G′ and G′′ increased significantly, and became less dependent on ω, which indicated that PLA/COP blends showed increased solid-like behavior. It is noted that an apparent plateau appeared in low frequency range of both G′–ω and G′′–ω plots when the content of COP increased from 10 wt% to 20 wt%. The plateau became more obvious with further increasing COP content to 30 wt%.
Generally, G′ is related to the elasticity of microstructure and G′′ is related to the molecular mobility in the melt. The enhancement in G′ represents improved elastic response of the melt under shear conditions, i.e., the melt exhibits the solid-like response. Similarly, the increase in G′′ indicated reduced molecular chain mobility. In this study, the formed COP in PLA matrix is a crosslinking polymer, which should show solid-like behavior, due to the frozen chain mobility by the formation of network structure. Therefore, the increase in COP content would reasonably reduce the chain mobility and enhance solid-like response of the blends by increasing the fraction of crosslinking parts of the blends.
Variation of loss tangent (tanδ) during frequency sweep provided another evidence for the formation of cross-linked COP network in PLA matrix. The frequency independence of tanδ, as reported by Winter and Chambon, could be used to indicate the gel point for cross-linking systems,43–45 and was also widely used to evaluate the percolation thresholds of polymer composites.46–48 Fig. 4d shows the frequency dependences of tanδ at 180 °C for neat PLA and PLA/COP blends with different compositions. For neat PLA, tanδ almost decreased with increasing frequency, which is a typical terminal behavior of a liquid-like material. PLA/COP blends showed much less significant frequency dependence of tanδ, and the tanδ decreased gradually with increase in COP content at low frequency range. It is obvious that a plateau at low frequency range of the tanδ–ω plot could be observed when the content of COP increased to 10 wt%, which indicated that a transition from liquid-like to gel-like behavior occurred, suggesting the presence of a threshold COP content for the network formation.
3.3. Thermal properties and crystallization behaviors
DSC was used to study the thermal transition and crystallization behaviors of neat PLA and PLA/COP blends. Fig. 5a shows the DSC heating scans of neat PLA and PLA/COP blends, and Table 1 summarizes the data obtained from the DSC analysis. Neat PLA showed a glass transition temperature (Tg) of 62.4 °C. The Tg of isolated COP was determined to be −6.5 °C, the DSC curve of COP was not shown for brevity. For polymer blends, the change of Tg with composition can be used to evaluate the miscibility between the blending components. It was found that the Tgs of PLA component in PLA/COP blends were very close to neat PLA. However, the values were somewhat lower than that of neat PLA and increased gradually with increasing COP content. The results indicated that PLA/COP blends showed limited miscibility and the miscible extent decreased with increasing COP content, which was in agreement with the results obtained by SEM observation, where the size of dispersed COP increased significantly with COP content. | ||
| Fig. 5 DSC heating scans of neat PLA and PLA/COP blends (a) and POM images for neat PLA (b), PLA/COP-5 (c), PLA/COP-10 (d), PLA/COP-20 (e), and PLA/COP-30 (f). | ||
| Sample | Tg (°C) | Tcc (°C) | ΔHcc (J g−1) | Tm (°C) | ΔHm (J g−1) |
|---|---|---|---|---|---|
| Neat PLA | 62.4 | 128.8 | 10.6 | 168.4 | 10.9 |
| PLA/COP-5 | 59.8 | 112.4 | 32.1 | 169.9 | 33.8 |
| PLA/COP-10 | 61.2 | 112.3 | 31.3 | 169.7 | 32.7 |
| PLA/COP-20 | 61.7 | 110.2 | 26.8 | 170.0 | 28.5 |
| PLA/COP-30 | 62.2 | 111.3 | 22.5 | 170.2 | 23.7 |
Neat PLA showed a broad cold crystallization exothermic peak with a respective cold crystallization temperature (Tcc) and enthalpy (ΔHcc) of 128.8 °C and 10.6 J g−1, indicating poor crystallizability. Surprisingly, after incorporation of COP, the cold crystallization exothermic peaks became very narrow and the Tccs shifted to much lower temperature compared to neat PLA, suggesting significantly improved crystallization. For the effect of COP content on the Tcc of PLA component, the value decreased first ant then increased with increasing COP content. The value of Tcc in the heating scan can be used to indicate the crystallization rate of polymers. Lower Tcc indicated faster crystallization rate. That is to say, the addition of COP increased the crystallization rate of PLA, which has positive significance for thermal processing of PLA. The Tcc values were 112.4, 112.3, 110.2 and 111.3 °C for the PLA component in PLA/COP-5, PLA/COP-10, PLA/COP-20 and PLA/COP-30, respectively. The ΔHcc values of PLA component in the blends were much higher than that of neat PLA, due to the improved crystallization. In addition, ΔHcc decreased gradually with increasing COP content, which was reasonable since the content of crystalline PLA component decreased. The increased crystallization rate of PLA by incorporation of COP may be ascribed to the increased nucleation effect of the blends, in which the interface of the phase separated domains may provide favorable nucleation sites for crystallization of PLA. Consequently, the crystallization rate of PLA was increased by decreasing the nucleation barrier in the blends. Similar results were also found in some other phase-separated PLA based blends.49,50
To confirm if nucleation effect increased by incorporation of COP, the spherulitic morphologies of neat PLA and PLA/COP blends were observed by POM after isothermally crystallized at 120 °C for 1 h, as shown in Fig. 5. Neat PLA (Fig. 5b) showed regular spherulites with very large diameters. The regularity of spherulites of PLA component in the blends decreased, and some particle-like impurities, COP dispersed domains, were observed within the spherulites. Furthermore, the nuclei densities of the spherulites in the blends were much higher than that in neat PLA, and increased with increasing COP content (Fig. 5c–f). Therefore, the enhanced crystallization rate of PLA component in the blends were reasonably attributed to the improved nucleation effect by formation of phase-separated PLA/COP blends.
Neat PLA showed a single melting peak with a melting point of 168.4 °C. The PLA/COP blends showed double melting peaks, which should be ascribed to the mechanism of melting, recrystallization, and remelting behaviors of the sample.51 The first melting peak at ∼163 °C should be attributed to the melting of the crystals formed in the cold crystallization stage during heating and the second melting peak should be ascribed to the melting of the crystals formed by reorganization of crystals at higher temperatures during heating process. It was found that the temperature of the second melting peak was ∼170 °C for all blends, which is slightly higher than that of neat PLA, due to the absence of recrystallization of neat PLA at high temperature during heating process.
Thermal stability of neat PLA, isolated COP and PLA/COP blends were investigated by TGA. Fig. 6 shows the TGA and DTG curves of the sample in the range of 50–700 °C at a heating rate of 10 °C min−1 under N2 atmosphere. Neat PLA showed a single stage decomposition behavior with an initial thermal decomposition temperature (T5, a temperature of 5% weight loss) of 348 °C and a maximum decomposition temperature (Tmax, corresponding to a maximum decomposition rate temperature) of 383 °C. The isolated COP showed a two-stage decomposition behavior with T5 of 325 °C. The first decomposition stage with Tmax of 350 °C was attributed to the urethane bond breaking and the second stage with Tmax of 420 °C was ascribed to the decomposition of castor oil soft segment of COP.37,39 All PLA/COP blends showed two-stage decomposition behaviors. The weight loss of the first stage decreased and that of the second stage increased with increasing COP content. What important is the effect of the added COP on the thermal stability of the blends. The T5 decreased from 348 °C of neat PLA to 343, 343, 330, and 325 °C for PLA/COP-5, PLA/COP-10, PLA/COP-20 and PLA/COP-30, respectively, due to the increasing content of COP with relatively lower T5. Although the initial thermal decomposition temperature decreased, the values were still much higher than that of processing temperature of PLA. That is to say, the blends were thermally stable during thermal processing. In addition, we could find that the final char residue increased with COP content due to its high inherent car residue.
 | ||
| Fig. 6 TGA (a) and DTG (b) curves of neat PLA, isolated COP, and PLA/COP blends from room temperature to 700 °C under N2 atmosphere. | ||
3.4. Mechanical properties
The tensile properties of neat PLA and PLA/COP blends were tested at room temperature by a universal testing machine. Fig. 7 shows the stress–strain curves of neat PLA and the PLA/COP blends. Neat PLA showed a brittle fracture behavior. Although a distinct yield point is observed, the sample broke immediately after yielding due to the instability of the formed neck. In contrast, all PLA/COP blends exhibited ductile character with clear yield point and stable neck growth during cold drawing. They were all broken at a very large elongation.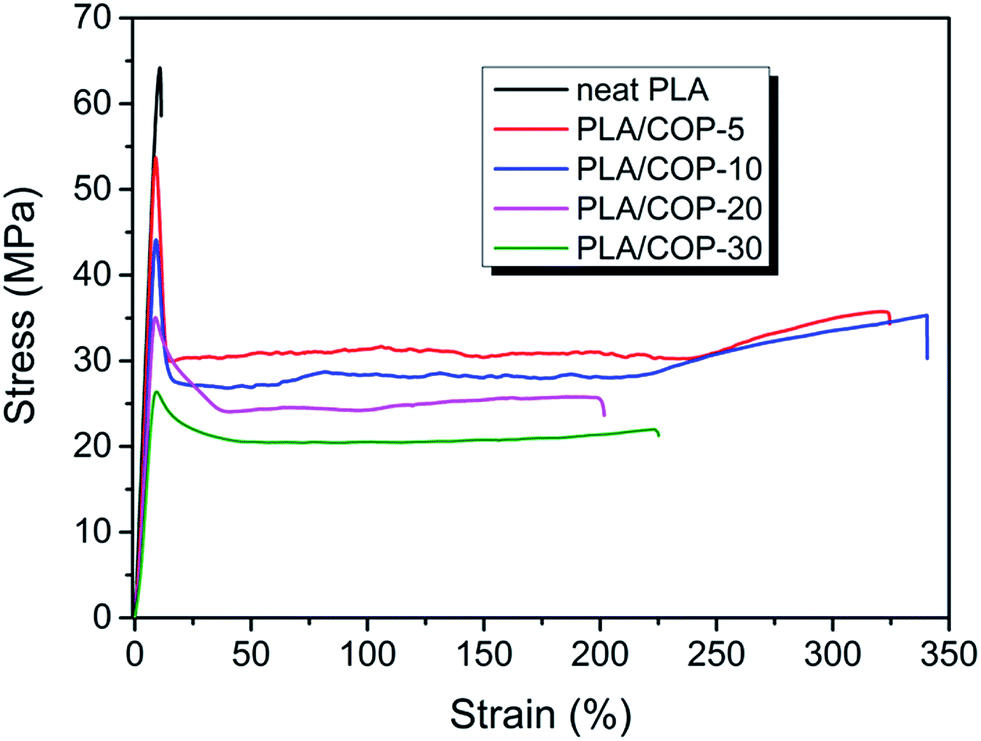 | ||
| Fig. 7 Stress–strain curves of neat PLA and PLA/COP blends with different compositions. | ||
Fig. 8 summarizes the data for the mechanical properties of neat PLA and PLA/COP blends with different compositions. The elongation at break, tensile strength and Young's modulus of rigid and brittle neat PLA were 7.5%, 65.5 MPa and 1.99 GPa, respectively. For PLA/COP blends, it is surprise to find that the elongation at break of the blend with only 5 wt% COP was increased by 45 times to 338% compared to neat PLA, whereas the tensile strength and Young's modulus were only reduced by ∼20% and 10% with the values of 52.39 MPa and 1.80 GPa, respectively. The increment in elongation at break was more than most of other castor oil based polymers toughened PLA blends with same composition, the elongation at break of PLA blends with 5 wt% of other castor oil based polymers were usually less than 50%.14,35,37,38 For the effect of COP content on the mechanical properties of the blends, the tensile strength and Young's modulus decreased gradually with increasing COP content due to its relative lower rigidity; while the elongation at break increased first and then decreased with increasing COP content. The maximum elongation at break occurred for the blend with 10 wt% COP. The reduced elongation at break of the blends with COP content more than 10 wt% could be attributed to the large size of COP and the poor interfacial adhesion between PLA and COP, as confirmed by SEM in the previous section that the COP size was greater than 10 μm for the blends with COP content ≥20 wt%.
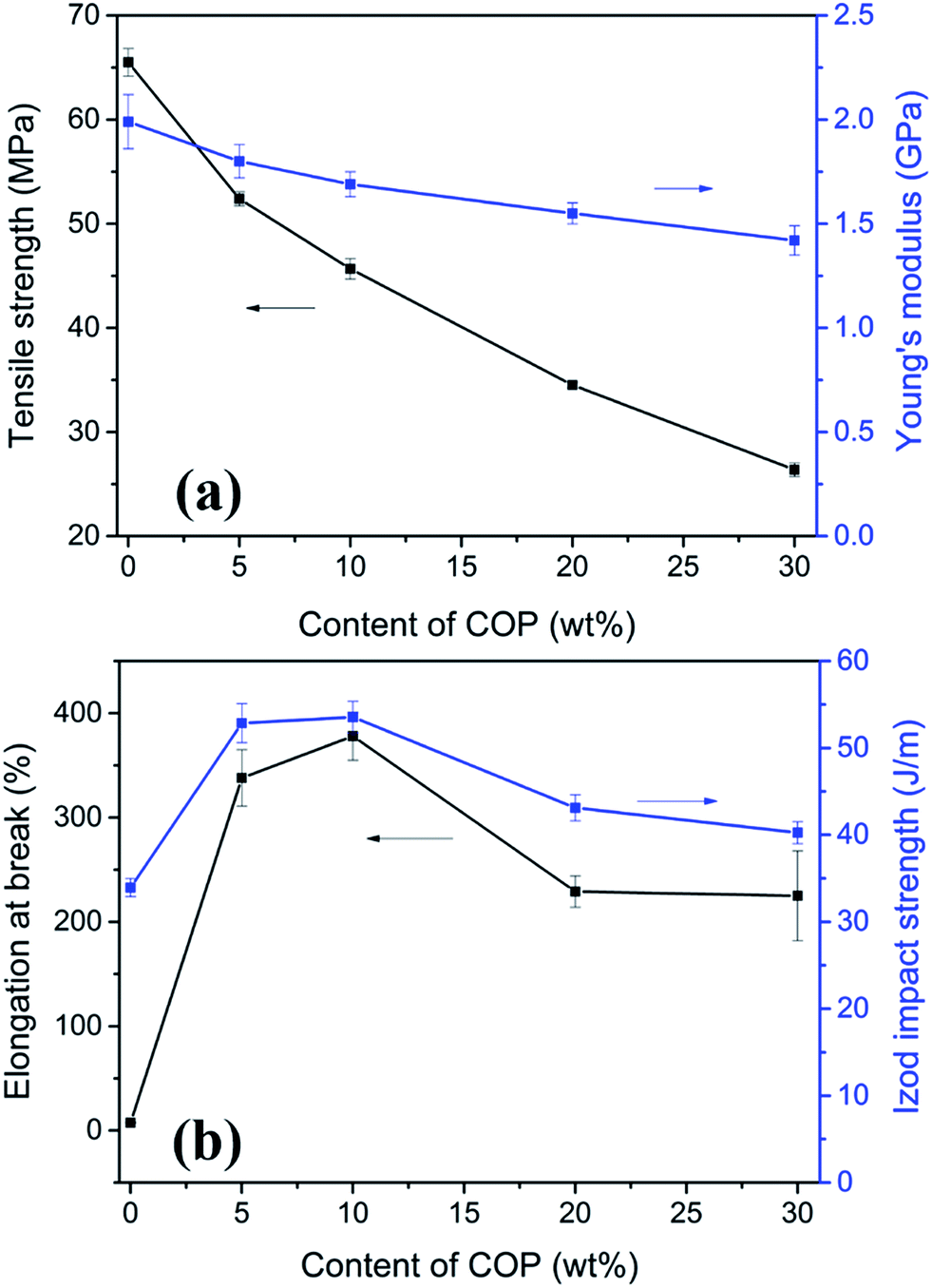 | ||
| Fig. 8 Mechanical properties of neat PLA and PLA/COP blends. (a) Tensile strength and Young's modulus, (b) elongation at break and notched Izod impact strength. | ||
The notched Izod impact strengths of PLA/COP blends were also measured and summarized in Fig. 8. The value showed similar variation tendency to the elongation at break versus the content of COP. The impact strength of neat PLA was 33.9 J m−1. Addition of 5 wt% COP increased the impact strength to 52.8 J m−1. The maximum impact strength was observed for the blend with 10 wt% COP, which showed an improvement of ∼58% compared to neat PLA. When the content of COP increased to 20 and 30 wt%, the impact strength decreased to 43.1 and 40.2 J m−1, respectively. The decrease in impact strength of the blends with high COP content should also related to the poor morphology of the blend with large COP dispersed phase size.
To further observe the insight into the effect of COP content on the tensile properties of the blends, we observed the morphology for the fractured surfaces of PLA/COP blends with different compositions, as shown in Fig. 9. Brittle neat PLA showed smooth surface without plastic deformation, for brevity, the SEM image of neat PLA was not shown. PLA/COP-5 (Fig. 9a) and PLA/COP-10 (Fig. 9b) showed very rough surfaces with significant plastic deformation occurred, corresponding to the large elongation at break during tensile testing. A lot of cavities were observed for the surfaces of PLA/COP-5 and PLA/COP-10, and particle debonding of dispersed COP phase from PLA matrix were also observed. Those cavities were formed during tension when the stress was higher than interfacial adhesion between PLA matrix and COP inclusions. Dispersed COP phase debonded from PLA matrix at the interface, then cavities grew, inducing large matrix plastic deformation. So, matrix plastic deformation caused by debonding induced shear yielding should be the toughening mechanism of the PLA/COP blends.52–55 It is obvious that the average size of cavities for PLA/COP-10 was greater than that for PLA/COP-5, due to the relatively smaller size of COP particle in PLA/COP-5, in which plenty of COP particles with size of submicron existed, as discussed in the above text. When the content of COP increased to 20 wt% (Fig. 9c) and 30 wt% (Fig. 9d), the blend samples also showed rougher surface compared to neat PLA but the extent of plastic deformation reduced largely with increasing COP content, corresponding to decreased elongation at break but greater than neat PLA. The large scaled COP particles with size greater than 10 μm would cause large cavities during debonding. The large cavities were not stable during tension thus broke before significant matrix plastic deformation developed, which should be responsible for the reduced elongation at break of PLA/COP blends with higher COP content.
 | ||
| Fig. 9 SEM images for tensile fractured surfaces of PLA/COP-5 (a), PLA/COP-10 (b), PLA/COP-20 (c), and PLA/COP-30 (d). | ||
4. Conclusions
This work demonstrated that dynamic vulcanization is applicable in fabrication of toughened PLA blends with vulcanized biomass-derived castor oil polyurethane, which was quickly formed during the process by crosslinking of castor oil with MDI. Phase separation occurred in the present PLA/COP dynamic vulcanization system and the size of dispersed COP domains increased from submicron to greater than 10 μm with COP content increased from 5 to 30 wt%. All the rheological parameters including melt viscosity, storage modulus, loss modulus and loss tangent increased significantly with increase in COP content, and a transition from liquid-like to gel-like response occurred when 10 wt% COP was incorporated. The PLA/COP blends showed good thermal stabilities with the initiation thermal decomposition temperature higher than 325 °C irrespective of compositions. The crystallization rate of PLA was increased by enhanced nucleation initiated from the interface of the phase separated domains of the immiscible PLA/COP blends. The dynamically vulcanized COP showed significant toughening efficiency toward PLA, as the elongation at break of PLA was increased by 43 times to 338% with the COP content of only 5 wt%, and the mechanical strength and modulus were only reduced very slightly. The toughening mechanism corresponded to cavitation induced matrix plastic deformation, where the cavitation was a result of particle debonding of COP from PLA matrix. The large-sized cavities, formed with increasing COP content due to the increased size of dispersed COP phase, showed relatively low stability during tension, which accounted for the relatively lower elongation at break of the blends with higher COP contents. The impact strength of PLA was increased by ∼58% with addition of 10 wt% COP, compared to neat PLA. In a word, dynamic vulcanization of castor oil in the presence of MDI within PLA matrix was successfully performed, which was proved to be a facial and cost-effective method to toughen PLA with biomass-derived castor oil derivatives.Acknowledgements
This work was supported by National Natural Science Foundation of China (51673158), the Opening Project of State Key Laboratory of Polymer Materials Engineering (Sichuan University) (Grant No. sklpme2015-4-26), and Fundamental Research Funds for the Central Universities (XDJK2015C022 and SWU115006).References
- J. B. Zeng, L. Jiao, Y. D. Li, M. Srinivasan, T. Li and Y. Z. Wang, Carbohydr. Polym., 2011, 83, 762–768 CrossRef CAS.
- M. M. Reddy, S. Vivekanandhan, M. Misra, S. K. Bhatia and A. K. Mohanty, Prog. Polym. Sci., 2013, 38, 1653–1689 CrossRef CAS.
- Y. S. He, J. B. Zeng, G. C. Liu, Q. T. Li and Y. Z. Wang, RSC Adv., 2014, 4, 12857–12866 RSC.
- H. Bai, H. Xiu, J. Gao, H. Deng, Q. Zhang, M. Yang and Q. Fu, ACS Appl. Mater. Interfaces, 2012, 4, 897–905 CAS.
- D. Garlotta, J. Polym. Environ., 2001, 9, 63–84 CrossRef CAS.
- K. S. Anderson, K. M. Schreck and M. A. Hillmyer, Polym. Rev., 2008, 48, 85–108 CrossRef CAS.
- H. Y. Yin, X. F. Wei, R. Y. Bao, Q. X. Dong, Z. Y. Liu, W. Yang, B. H. Xie and M. B. Yang, ACS Sustainable Chem. Eng., 2015, 3, 654–661 CrossRef CAS.
- Z. Liu, Y. Luo, H. Bai, Q. Zhang and Q. Fu, ACS Sustainable Chem. Eng., 2016, 4, 111–120 CrossRef CAS.
- R. M. Rasal, A. V. Janorkar and D. E. Hirt, Prog. Polym. Sci., 2010, 35, 338–356 CrossRef CAS.
- H. Liu and J. Zhang, J. Polym. Sci., Part B: Polym. Phys., 2011, 49, 1051–1083 CrossRef CAS.
- Y. B. Wang and M. A. Hillmyer, J. Polym. Sci., Part A: Polym. Chem., 2001, 39, 2755–2766 CrossRef CAS.
- H. I. Oyama, Polymer, 2009, 50, 747–751 CrossRef CAS.
- Z. Su, Q. Li, Y. Liu, G.-H. Hu and C. Wu, Eur. Polym. J., 2009, 45, 2428–2433 CrossRef CAS.
- M. L. Robertson, J. M. Paxton and M. A. Hillmyer, ACS Appl. Mater. Interfaces, 2011, 3, 3402–3410 CAS.
- H. Kang, B. Qiao, R. Wang, Z. Wang, L. Zhang, J. Ma and P. Coates, Polymer, 2013, 54, 2450–2458 CrossRef CAS.
- T. Lebarbe, E. Grau, B. Gadenne, C. Alfos and H. Cramail, ACS Sustain. Chem. Eng., 2015, 3, 283–292 CrossRef CAS.
- G. Coativy, M. Misra and A. K. Mohanty, ACS Sustain. Chem. Eng., 2016, 4, 2142–2149 CrossRef CAS.
- S. C. Mauck, S. Wang, W. Ding, B. J. Rohde, C. K. Fortune, G. Yang, S. K. Ahn and M. L. Robertson, Macromolecules, 2016, 49, 1605–1615 CrossRef CAS.
- N. Bitinis, R. Verdejo, P. Cassagnau and M. A. Lopez-Manchado, Mater. Chem. Phys., 2011, 129, 823–831 CrossRef CAS.
- J. B. Zeng, K. A. Li and A. K. Du, RSC Adv., 2015, 5, 32546–32565 RSC.
- H. Liu, F. Chen, B. Liu, G. Estep and J. Zhang, Macromolecules, 2010, 43, 6058–6066 CrossRef CAS.
- H. Liu, W. Song, F. Chen, L. Guo and J. Zhang, Macromolecules, 2011, 44, 1513–1522 CrossRef CAS.
- Y. Chen, D. Yuan and C. Xu, ACS Appl. Mater. Interfaces, 2014, 6, 3811–3816 CAS.
- G. C. Liu, Y. S. He, J. B. Zeng, Q. T. Li and Y. Z. Wang, Biomacromolecules, 2014, 15, 4260–4271 CrossRef CAS PubMed.
- R. R. Babu and K. Naskar, in Advanced Rubber Composites, ed. G. Heinrich, 2011, vol. 239, pp. 219–247 Search PubMed.
- D. Yuan, Z. Chen, C. Xu, K. Chen and Y. Chen, ACS Sustain. Chem. Eng., 2015, 3, 2856–2865 CrossRef CAS.
- Y. Chen, K. Chen, Y. Wang and C. Xu, Ind. Eng. Chem. Res., 2015, 54, 8723–8731 CrossRef CAS.
- Y. Wang, K. Chen, C. Xu and Y. Chen, J. Phys. Chem. B, 2015, 119, 12138–12146 CrossRef CAS PubMed.
- D. Yuan, C. Xu, Z. Chen and Y. Chen, Polym. Test., 2014, 38, 73–80 CrossRef CAS.
- D. Yuan, K. Chen, C. Xu, Z. Chen and Y. Chen, Carbohydr. Polym., 2014, 113, 438–445 CrossRef CAS PubMed.
- P. Ma, P. Xu, Y. Zhai, W. Dong, Y. Zhang and M. Chen, ACS Sustain. Chem. Eng., 2015, 3, 2211–2219 CrossRef CAS.
- X. Lu, X. Wei, J. Huang, L. Yang, G. Zhang, G. He, M. Wang and J. Qu, Ind. Eng. Chem. Res., 2014, 53, 17386–17393 CrossRef CAS.
- H. Kang, X. Hu, M. Li, L. Zhang, Y. Wu, N. Ning and M. Tian, RSC Adv., 2015, 5, 23498–23507 RSC.
- K. R. Kunduru, A. Basu, M. Haim Zada and A. J. Domb, Biomacromolecules, 2015, 16, 2572–2587 CrossRef CAS PubMed.
- S. Huang, H. Sun, J. Sun, G. Li and X. Chen, Mater. Lett., 2014, 133, 87–90 CrossRef CAS.
- T. Lebarbe, E. Ibarboure, B. Gadenne, C. Alfos and H. Cramail, Polym. Chem., 2013, 4, 3357–3369 RSC.
- T. Gurunathan, S. Mohanty and S. K. Nayak, J. Mater. Sci., 2014, 49, 8016–8030 CrossRef CAS.
- J. Fujigasaki and M. Shibata, J. Polym. Res., 2015, 22, 215 CrossRef.
- E. Hablot, D. Zheng, M. Bouquey and L. Averous, Macromol. Mater. Eng., 2008, 293, 922–929 CrossRef CAS.
- Y. D. Li, J. B. Zeng, X. L. Wang, K. K. Yang and Y. Z. Wang, Biomacromolecules, 2008, 9, 3157–3164 CrossRef CAS PubMed.
- G. C. Liu, Y. S. He, J. B. Zeng, Y. Xu and Y. Z. Wang, Polym. Chem., 2014, 5, 2530–2539 RSC.
- P. Juntuek, C. Ruksakulpiwat, P. Chumsamrong and Y. Ruksakulpiwat, J. Appl. Polym. Sci., 2012, 125, 745–754 CrossRef CAS.
- H. Fang, F. Jiang, Q. Wu, Y. Ding and Z. Wang, ACS Appl. Mater. Interfaces, 2014, 6, 13552–13563 CAS.
- H. H. Winter and F. Chambon, J. Rheol., 1986, 30, 367–382 CrossRef CAS.
- F. Chambon and H. H. Winter, J. Rheol., 1987, 31, 683–697 CrossRef CAS.
- C. Liu, J. Zhang, J. He and G. Hu, Polymer, 2003, 44, 7529–7532 CrossRef CAS.
- W. Li, Y. Zhang, J. Yang, J. Zhang, Y. Niu and Z. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 6468–6478 CAS.
- Y. Pan and L. Li, Polymer, 2013, 54, 1218–1226 CrossRef CAS.
- R. Dell'Erba, G. Groeninckx, G. Maglio, M. Malinconico and A. Migliozzi, Polymer, 2001, 42, 7831–7840 CrossRef.
- J. Lu, Z. Qiu and W. Yang, Polymer, 2007, 48, 4196–4204 CrossRef CAS.
- Y. T. Shieh and G. L. Liu, J. Polym. Sci., Part B: Polym. Phys., 2007, 45, 1870–1881 CrossRef CAS.
- L. Jiang, M. P. Wolcott and J. W. Zhang, Biomacromolecules, 2006, 7, 199–207 CrossRef CAS PubMed.
- J. J. Han and H. X. Huang, J. Appl. Polym. Sci., 2011, 120, 3217–3223 CrossRef CAS.
- V. Ojijo, S. S. Ray and R. Sadiku, ACS Appl. Mater. Interfaces, 2013, 5, 4266–4276 CAS.
- P. Ma, X. Cai, Y. Zhang, S. Wang, W. Dong, M. Chen and P. J. Lemstra, Polym. Degrad. Stab., 2014, 102, 145–151 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2016 |