
The excellent photocatalytic synergism of PbBiO2Br/UiO-66-NH2 composites via multiple coupling effects
Sunfeng Li,
Xing Wang,
Yanli Xu,
Hanbiao Yang,
Fengyu Wei* and
Xueting Liu*
School of Chemistry and Chemical Engineering, Hefei University of Technology, Anhui Key Laboratory of Controllable Chemical Reaction & Material Chemical Engineering, Hefei 230009, Anhui, China. E-mail: weifyliuj@163.com; wmlxt@163.com
First published on 14th September 2016
Abstract
PbBiO2Br/UiO-66-NH2 composites synthesized by a facile solvothermal method have a core–shell structure with UiO-66-NH2 forming the shell around a PbBiO2Br core. Their photocatalytic activities in the degradation of Rhodamine B (RhB) dye solution and colorless phenol solution under visible light irradiation were investigated. For both model organic pollutants, the PbBiO2Br/UiO-66-NH2 composites exhibit higher photocatalytic activity than the pure components. This synergistic effect is due to the high adsorption capacity of UiO-66-NH2 and the compounding induced an enhanced separation efficiency of photogenerated electron–hole pairs. The composite with a 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 mass ratio of PbBiO2Br to ZrCl4 exhibits the highest photocatalytic activity for RhB degradation, in which trapping experiments confirm that the photogenerated holes and O2˙− radicals are the main active species.
:2 mass ratio of PbBiO2Br to ZrCl4 exhibits the highest photocatalytic activity for RhB degradation, in which trapping experiments confirm that the photogenerated holes and O2˙− radicals are the main active species.
1. Introduction
In recent years, dyes have been widely used in the textile, paper and printing industries, so the treatment of dye-containing wastewater is a crucial problem to solve.1–3 Semiconductor (SC) photocatalysis for the photodegradation of organic dyes in water has emerged as a renewable technology.4–6Up until now, numerous materials which show photocatalytic activity under visible light irradiation have been reported.7–11 PbBiO2Br has aroused a growing interest recently, due to its band gap of 2.47 eV and unique layered structure.12,13 The assembled PbBiO2Br nanosheets show excellent photocatalytic activity for organic contaminant degradation under visible light irradiation, which is primarily attributed to the structure and the low recombination rate of charges in the ultrathin nanosheets.14
Recent research has been focused on metal–organic frameworks (MOFs), which are made of metal clusters linked by organic ligands.15–19 Due to their uniform but tunable pore size and high specific surface area, MOFs are considered for adsorption,20 storage,21–23 and health care applications.24 A group at Valencia25 first synthesized a zirconium(IV)-based MOF (UiO-66-NH2), which is based on a Zr6O4(OH)4 octahedron and a lattice formed by 12-fold connection through a 2-amino-1,4-benzene-dicarboxylate linker. The UiO-66-NH2 framework is robust and can undergo isoreticular functionalization without losing its high hydrothermal and chemical stability.26 Shen et al. have shown that UiO-66-NH2 has high visible light photocatalytic activity for reducing Cr(VI).27
It has been found that the combination of two different semiconductors can yield an enhanced photocatalytic activity via a synergistic effect such as more efficient charge separation.28,29 In this work, PbBiO2Br/UiO-66-NH2 composites prepared by a facile solvothermal method exhibit enhanced photocatalytic activity for degradation of both RhB dye solution and colorless phenol solution under visible light by its comparison with the pure components.
2. Experimental
2.1 Materials
Bismuth nitrate pentahydrate, ethanol, lead nitrate, hexadecyl trimethyl ammonium bromide (CTAB), ammonia water, benzoic acid, zirconium tetrachloride, N,N-dimethyl formamide (DMF) and chloroform were supplied from Sinopharm Chemical Reagent Co., Ltd. 2-Amino-1,4-benzenedicarboxylic acid was purchased from Tokyo Chemical Industry Co., Ltd. And all of chemicals were used as received without further purification.2.2 Preparation of catalysts
Assembly ultrathin PbBiO2Br nanosheets samples were synthesized using a solvothermal method.30 In a typical procedure, 0.5 mmol Bi(NO3)3·5H2O was added into 20 mL of ethanol containing stoichiometric amounts of hexadecyl trimethyl ammonium bromide (CTAB) and Pb(NO3)2 with continuous stirring, and then 5 mL ammonia water was added into this solution. The mixture solution was stirred for at least 30 min and then poured into a 50 mL Teflon-lined stainless autoclave. The autoclave was heated at 180 °C for 12 h under autogenous pressure and then cooled to room temperature. The resulting precipitates were collected and washed with ethanol and deionized water thoroughly and dried at 70 °C in air.The PbBiO2Br/UiO-66-NH2 composites were synthesized as UiO-66-NH2 with different PbBiO2Br/ZrCl4 ratios.25 A typical method is as follows: ZrCl4 (0.0848 g, 0.36 mmol), 2-amino-1,4-benzenedicarboxylic acid (0.0656 g, 0.036 mmol) and benzoic acid (0.984 g, 8.0 mmol) were dissolved in DMF (42 mL). PbBiO2Br (0.127 g, 0.24 mmol) was added into the solution under ultrasonic vibration for 30 min. The mixture was transferred to a stainless steel Teflon-lined autoclave of 50 mL capacity and then maintained at 393 K for 24 h. Then, the autoclave was cooled in air to room temperature, and the resulting solid was filtered, repeatedly washed with CHCl3 and dried at room temperature. The prepared composite is called PbBiO2Br/UiO-66-NH2 (3:2) where the mass ratio of PbBiO2Br to ZrCl4 is 3:2. Similarly, when the mass ratio of PbBiO2Br to ZrCl4 is x:y, the composite is named PbBiO2Br/UiO-66-NH2 (x:y).
2.3 Characterization
The morphological analysis of the samples was done using a JEM-2100F field emission transmission electron microscopy (FETEM) equipped with an energy-dispersive X-ray spectrometer (EDS). X-ray diffraction (XRD) patterns of the samples were determined in the range of 2θ = 4–60° on a Rigaku D/max-2500V X-ray diffractometer using Cu-Kα (λ = 0.154 nm) radiation. N2 adsorption–desorption (BET) was performed on a Tristar II 3020M surface area and porosity analyzer at 77 K. Before the actual measurements, the sample was degassed at 70 °C for 3 h. X-ray photoelectron spectroscopy (XPS) was conducted using an ESCALAB250 spectrometer. UV-vis spectra were recorded on a DUV-3700 spectrometer. Photoluminescence emission spectra (PL) were measured on a PL measurement system (Fluorolog Tau-3) with the excitation wavelength of 320 nm.2.4 Adsorption experiments
The adsorption of dye was measured at ambient pressure and 298 K in the dark. 20 mg of the photocatalyst was added into 100 mL dye aqueous solution (20 mg L−1) with continuous stirring. Samples were taken at a fixed time, and were filtered by a 0.22 μm filter. The absorbance of the filtrate was measured using a Shimadzu UV-240 spectrophotometer.2.5 Photocatalytic experiments
The photocatalytic degradation of dye was measured at ambient pressure and 298 K in a home made photochemical reaction equipment. The light source was a PHILIPS 70 W metal halide lamp (λ < 380 nm was filtered out by a cut off filter). 20 mg photocatalyst was added into 100 mL dye (5 mg L−1) aqueous solution. Before irradiation, the suspension was continuously stirred for 12 h in the dark in order to reach adsorption–desorption equilibrium between dye and the photocatalyst. The supernatant liquid was obtained by filtration using 0.22 μm filter and examined on a Shimadzu UV-240 spectrophotometer. For comparison, the photocatalytic activities of UiO-66-NH2 and PbBiO2Br were also tested under the same conditions. The degradation of colorless model pollutant phenol (5 mg L−1) was also conducted under the identical condition with RhB.3. Results and discussion
3.1 Morphology and composition of PbBiO2Br/UiO-66-NH2 photocatalysts
It can be seen from Fig. 1a and b that pure UiO-66-NH2 comprises disc-like particles with the particle size in the range of 30–40 nm. In PbBiO2Br/UiO-66-NH2(3:2), the UiO-66-NH2 particles are still clearly identifiable and the PbBiO2Br flakes can be also observed. Although the exact size of the PbBiO2Br flakes is difficult to estimate from the TEM images, the thickness of the flakes is within the range of 6 to 12 nm. And they exhibit a compact patter, as shown in the TEM image (Fig. 1c and d). The lattice fringes of the (101) plane of PbBiO2Br can be obviously seen in Fig. 1e from which the clear heterojunction interfaces of PBBiO2Br/UiO-66-NH2 are displayed. These indicate that the PbBiO2Br nanosheets microcrystallines are in intimate contact with UiO-66-NH2 particles.
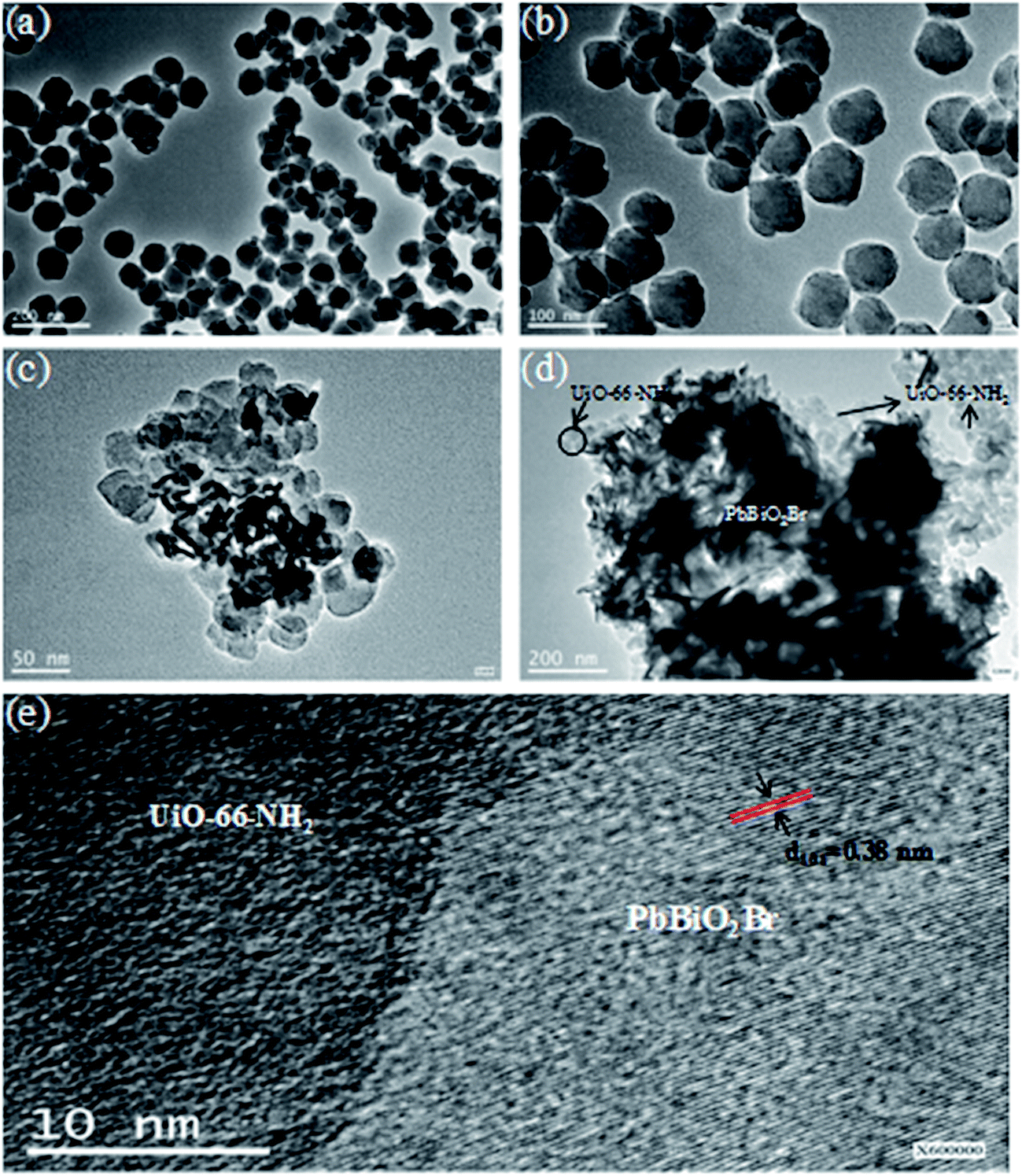 | ||
| Fig. 1 TEM images of (a and b) UiO-66-NH2, (c and d) PbBiO2Br/UiO-66-NH2 (3:2), and (e) HRTEM image of PbBiO2Br/UiO-66-NH2 (3:2). | ||
The EDS spectra of PbBiO2Br/UiO-66-NH2 (3:2) indicate that Br, Bi, Pb and Zr elements are the major chemical components present in the composite. It can be seen from Fig. 1c and 2d that PbBiO2Br and UiO-66-NH2 exist with the different configurations in the composite. The Br, Bi and Pb distributions by EDS mapping (Fig. 2a–c) show that the flakes are PbBiO2Br in the composite. However, the Zr elements are wholly distributed around the flakes as a shell (Fig. 2d). These confirm that the composite possesses a hierarchical core–shell structure with UiO-66-NH2 forming the shell around the PbBiO2Br core.
 | ||
| Fig. 2 (a) Br, (b) Bi, (c) Pb and (d) Zr distribution by EDS mapping of PbBiO2Br/UiO-66-NH2 (3:2). | ||
The XRD patterns of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2 (3:2) are shown in Fig. 3a, which are in accordance with the reported values.14,25,26 By comparing the XRD patterns of PbBiO2Br/UiO-66-NH2 (3:2) with those of the UiO-66-NH2 and PbBiO2Br precursors, it can be seen that the composite displays the characteristic peaks of both UiO-66-NH2 and PbBiO2Br, and the intensity and location of the peaks are changed somewhat. That is, the composite is not a simple physical mixture, and there exists interfacial interactions between UiO-66-NH2 and PbBiO2Br.
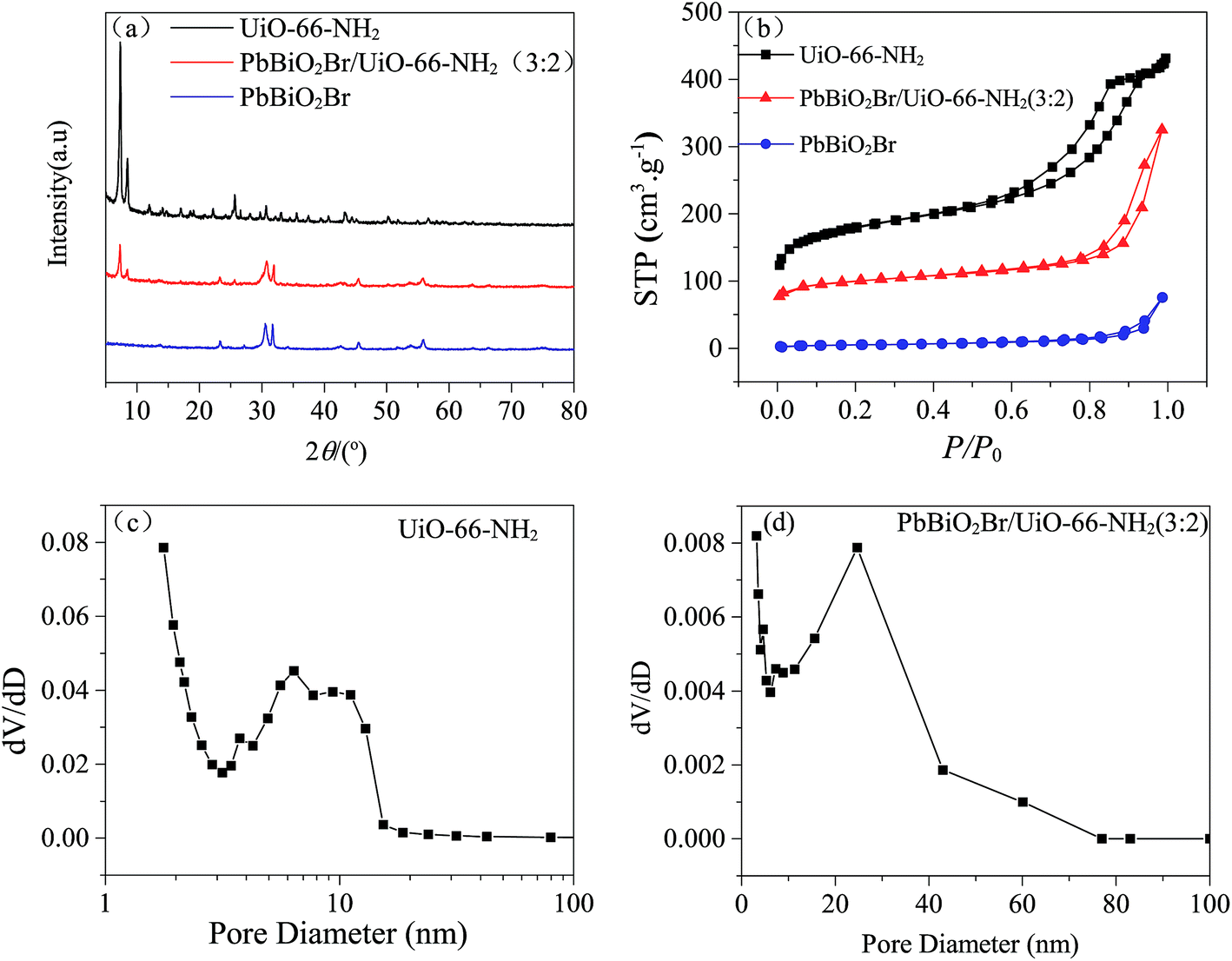 | ||
| Fig. 3 (a) XRD patterns of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2(3:2); (b) the N2 adsorption–desorption isotherms of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2 (3:2); (c) the BJH pore diameter distribution curve of UiO-66-NH2; (d) the BJH pore diameter distribution curve of PbBiO2Br/UiO-66-NH2 (3:2). | ||
The N2 adsorption–desorption isotherms of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2 (3:2) are displayed in Fig. 3b, and the values of BET surface area of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2 (3:2) are 616.55 m2 g−1, 18.14 m2 g−1 and 320.55 m2 g−1, respectively. The N2 isotherm of UiO-66-NH2 is categorized as type IV. This property implies the presence of mesopores (between 2 to 50 nm in size). As shown in Fig. 3c, the BJH pore size of UiO-66-NH2 is mainly distributed in the range of 6 to 12 nm, which is attributed to the agglomeration of particles. In contrast, there is no mesopores structure in PbBiO2Br. The surface area of PbBiO2Br/UiO-66-NH2 (3:2) is higher than that of pure PbBiO2Br, and its BJH pore diameter distribution is not as homogeneous as in UiO-66-NH2, but it has even larger pore size. The high surface area and large pore size can enhance the adsorption and reaction for dye molecules on surface active sites. This phenomenon favors for the enhancement in photocatalytic activity.
X-ray photoelectron spectroscopy (XPS) was carried out on the PbBiO2Br/UiO-66-NH2 (3:2) sample to determine the surface compositions and chemical states of the elements. The shift of the peak position on the charge effect was calibrated by using the binding energy of C 1s at 284.78 eV. The survey XPS spectra of PbBiO2Br/UiO-66-NH2 (3:2) sample are shown in Fig. 4a. Fig. 4b–f show high-resolution XPS spectra of the primary elements. The Br 3d5/2 and Br 3d3/2 peaks are associated with the binding energies at 68.8 and 69.57 eV (Fig. 3b). The asymmetric XPS peak of O 1s (Fig. 4c) indicates that oxygen species are present in the form of lattice oxygen and hydroxyl groups adhered onto the surface.18,31 Two peaks at 164.68 and 159.25 eV (Fig. 4d) are assigned to Bi 4f5/2 and Bi 4f7/2, respectively, which are assigned to Bi3+ in the composites.32 The Pb 4f7/2 and Pb 4f5/2 peaks are associated with the binding energies at 138.44 and 143.33 eV (Fig. 4e), respectively. The curves of Zr 3d region could be deconvoluted into two peaks for Zr 3d5/2 and Zr 3d3/2 locating at around 182.93 eV and 185.28 eV (Fig. 4f), respectively, which shift to the lower energy compared with the initial UiO-66-NH2.26 Therefore, it may be concluded that PbBiO2Br/UiO-66-NH2 (3:2) heterostructure photocatalysts have been successfully synthesized.
 | ||
| Fig. 4 XPS spectra (a) and high resolution Br 3d (b), O 1s (c), Bi 4f (d), Pb 4f (e) and Zr 3d (f) spectra of PbBiO2Br/UiO-66-NH2 (3:2). | ||
The UV-vis absorption spectra of PbBiO2Br, UiO-66-NH2 and PbBiO2Br/UiO-66-NH2 (3:2) were also studied (Fig. 5a). Nanosheet PbBiO2Br has a strong absorption in the visible range up to 500 nm,30 which is due to the intrinstic transition of the semiconductor. The steep shape rise that UiO-66-NH2 shows in the range of 300–500 nm is due to the band gap transition.26 Near the absorption band edge, the optical absorption has the following behavior:
| αhv = A(hv − Eg)n/2 | (1) |
 | ||
| Fig. 5 (a) UV-vis absorption spectra of UiO-66-NH2, PbBiO2Br and PbBiO2Br/UiO-66-NH2 (3:2); (b) PL spectra of UiO-66-NH2, PbBiO2Br, physical mixture and PbBiO2Br/UiO-66-NH2 (3:2). | ||
It is well known that PL (photoluminescence emission spectra) can be induced by the recombination between photogenerated electrons and holes. The lower the PL peak is, the less the recombination of electron–hole pairs is. Therefore, PL analysis is often performed to investigate the photogenerated charge separation efficiency. As shown in Fig. 5b, the PL emission spectra for samples under excitation at 320 nm were examined in the wavelength range of 340–800 nm, and the order of the PL spectra intensities is as following: UiO-66-NH2 > PbBiO2Br/UiO-66-NH2 (physical mixture) > PbBiO2Br/UiO-66-NH2 (3:2) > PbBiO2Br. As indicated, PbBiO2Br has very low PL spectrum intensity, whereas UiO-66-NH2 exhibits strong PL spectra. The PL spectrum intensity of PbBiO2Br/UiO-66-NH2 (3:2) is lower than that of the corresponding physical mixture, which means the separation of photogenerated electron–hole pairs is enhanced for the PbBiO2Br/UiO-66-NH2 (3:2) binary composite.
3.2 Adsorption activity of PbBiO2Br/UiO-66-NH2 photocatalysts
The adsorption performance of pure materials and composites was studied (Fig. 6). The adsorbed quantity, qe, can be calculated using the following relationship:
 | (2) |
 | ||
| Fig. 6 (a) Adsorption capacity of RhB onto PbBiO2Br, UiO-66-NH2, and PbBiO2Br/UiO-66-NH2 (3:2); (b) adsorption capacity of RhB on PbBiO2Br/UiO-66-NH2 composites. | ||
PbBiO2Br displays low adsorption capacity of RhB in contrast with UiO-66-NH2 and PbBiO2Br/UiO-66-NH2 (3:2) (Fig. 6a). This is due to the big BET surface area and the mesopores structure of UiO-66-NH2 and the electrostatic attraction interaction between RhB and UiO-66-NH2.34 Meanwhile, the adsorption of RhB onto UiO-66-NH2 has a great advantage as compared with that onto inorganic metal oxides such as PbBiO2Br, because there exists various interactions like pi–pi stacking, hydrogen bonding, etc., between aromatic rings of RhB and UiO-66-NH2. As shown in Fig. 6a and b, the adsorption activities of the binary composites are between those of PbBiO2Br and UiO-66-NH2, and increase with the increasing of UiO-66-NH2 content.
3.3 Photocatalytic activity of UiO-66-NH2, PbBiO2Br and the composites
The photocatalytic activities of UiO-66-NH2, PbBiO2Br and the composites were evaluated using the degradation of RhB under visible light irradiation. Owing to small BET surface area of PbBiO2Br and poor separation efficiency of photogenerated electron–hole pairs of UiO-66-NH2, both PbBiO2Br and UiO-66-NH2 show low photocatalytic activity before compounding, as shown in Fig. 7a. However, the PbBiO2Br/UiO-66-NH2 composites exhibit a greatly enhanced photocatalytic activity, which may be due to the result of the combination of two different semiconductors. | ||
| Fig. 7 (a) RhB degradation over various photocatalysts; (b) absorption spectra of RhB after irradiation times in the presence of PbBiO2Br/UiO-66-NH2 (3:2) composite; (c) photodegradation of phenol (5 mg L−1) on PbBiO2Br, UiO-66-NH2, and PbBiO2Br/UiO-66-NH2 (3:2). | ||
The photocatalytic activities of PbBiO2Br/UiO-66-NH2 with differing PbBiO2Br/ZrCl4 ratios were studied, and the results are displayed in Fig. 7a. All the composites exhibit a higher photocatalytic activity than two pure components. In particular, the composite with the PbBiO2Br/ZrCl4 ratio at 3:2 displays the highest photocatalytic activity, and PbBiO2Br/UiO-66-NH2 (3:2) exhibits a higher photocatalytic activity than the mechanically mixed PbBiO2Br/UiO-66-NH2 (3:2) composites, which indicates that the synergistic effect between PbBiO2Br and UiO-66-NH2 is the best.34 As shown in Fig. 7b, the absorption of RhB in the visible light region significantly decreased in the presence of PbBiO2Br/UiO-66-NH2 (3:2) composite. From this figure, the decrease of the characteristic absorption band of RhB at 552 nm can be obviously observed.
In addition, phenol is a colorless model organic pollutant, and the contribution of dye-sensitization during the degradation process could be ruled out. It was used as the second model pollutant to further evaluate the visible light photocatalytic performance of the as-prepared samples under visible light (λ < 380 nm was filtered out by a cut off filter), and the obtained results are illustrated in Fig. 7c. It is also indicated that the PbBiO2Br/UiO-66-NH2 (3:2) exhibits a higher photocatalytic activity than two pure components.
The regeneration of the photocatalyst is one of the important steps for practical applications. The stability of PbBiO2Br/ZrCl4 (3:2) was investigated, and after each photodegradation, it was separated from solution by centrifuge, and can be reused without considerable amount of mass loss. As shown in Fig. 8a, after five cycles, the K value stabilizes at about 0.245 min−1, which is 89.74% of the first cycle. The good structural stability of PbBiO2Br/ZrCl4 (3:2) was further verified by XRD, as shown in Fig. 8b.
 | ||
| Fig. 8 (a) Kinetics and rate constant of RhB photodegradation on the recycled PbBiO2Br/UiO-66-NH2 (3:2); (b) XRD patterns of PbBiO2Br/UiO-66-NH2 (3:2) before and after photocatalysis. | ||
3.4 Photodegradation mechanism of RhB
In order to evaluate the role of various active oxidants, scavengers were added to the photocatalytic system. These are tert-butyl alcohol (t-BuOH) for ˙OH,35 benzoquinone (BQ) for O2˙−36 and disodium ethylenediaminetetraacetate dehydrate (EDTA-2Na) for the holes h+.37–39The different active species trapping experiments for the degradation of RhB over the pure PbBiO2Br, UiO-66-NH2 and PbBiO2Br/UiO-66-NH2 (3:2) samples were first carried out to explore the enhancing photocatalytic mechanism. As shown in Fig. 9, for pure PbBiO2Br, when EDTA-2Na, BQ and t-BuOH were added into reaction solution, the corresponding degradation rates of RhB decrease, and maintain at 59.27%, 83.85% and 81.1% of that of pure PbBiO2Br, respectively, implying that the holes h+ are the major reactive species. In contrast, the photodegradation kinetics constants only change a little for all species trapping experiments for pure UiO-66-NH2. On the other hand, for the PbBiO2Br/UiO-66-NH2 (3:2) sample, when EDTA-2Na was added, the photocatalytic degradation rate decreases significantly, and only maintains at 22.45% of that without adding EDTA-2Na, indicating the holes h+ are the predominant active species. When the BQ was added into reaction solution, the degradation rate of RhB is inhibited slightly (43.58%), suggesting O2˙− also plays an important role in the photocatalytic process. When t-BuOH was added, the photocatalytic degradation rate basically keeps unchanged (85.63%), indicating that ˙OH plays a little role for the degradation of RhB. Therefore, the h+ and O2˙− radical are the major reactive species in the PbBiO2Br/UiO-66-NH2 (3:2) reaction system.
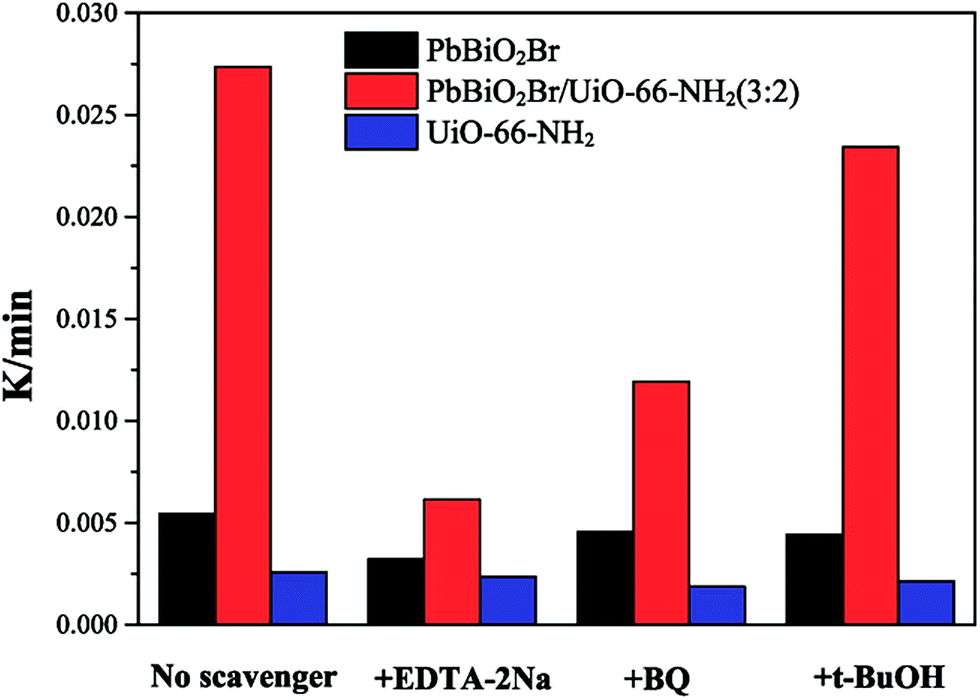 | ||
| Fig. 9 The species trapping experiments for degradation of RhB over pure PbBiO2Br, UiO-66-NH2 and PbBiO2Br/UiO-66-NH2 (3:2) photocatalysts under light irradiation. | ||
A photodegradation mechanism for the PbBiO2Br/UiO-66-NH2 composites under visible light irradiarion is shown in Fig. 10. The conduction bands (CB) of PbBiO2Br and UiO-66-NH2 are at −0.8 and −1.00,13,40 respectively, which are more negative than the standard redox potential of O2/O2˙− (−0.33 eV) and O2/HOO˙ (−0.037 eV).41 As a result, the photogenerated electrons in the CB of PbBiO2Br and UiO-66-NH2 can reduce O2 to give O2˙− or HOO˙, and the ˙OH radical can be generated from HOO˙.42 At the PbBiO2Br/UiO-66-NH2 heterojunction interface, the photogenerated electrons in the CB of UiO-66-NH2 are transferred to the CB of PbBiO2Br, and the holes from the valence band (VB) of PbBiO2Br are transferred to the VB of UiO-66-NH2. Therefore, UiO-66-NH2 can act as both an electron acceptor and donor. Hence, the electrons can easily migrate to the surface of PbBiO2Br and the redundant electrons on PbBiO2Br can also be transferred to UiO-66-NH2. As a result, the photogenerated electrons and holes are efficiently separated between PbBiO2Br and UiO-66-NH2. And the UiO-66-NH2 shell can enhance the adsorption of RhB cationic dye from the solution.43 Thereby enhances the photocatalytic activity. The photogenerated electrons in the CB of PbBiO2Br including those from the CB of UiO-66-NH2 can be captured by dissolved O2 to yield first the superoxide radical anion, O2˙−, then the HOO˙ radical upon protonation, and finally the ˙OH radical via trapping the electron.42 However, only small amount of the dye was oxidized by the ˙OH radical, and most of the dye was directly destroyed by the photogenerated holes in the VB of UiO-66-NH2 including those from the VB of PbBiO2Br. From Fig. 10, we can see that the electron–hole transfer at the PbBiO2Br/UiO-66-NH2 heterojunction interface enhances the separation of the electron–hole pairs.44 Furthermore, after bringing together PbBiO2Br and UiO-66-NH2, the resulting composites have the advantage of a high adsorption capacity especially for cationic dyes like RhB. These may be the reasons why the PbBiO2Br/UiO-66-NH2 composites have a synergistically enhanced photocatalytic performance as compared with the pure component materials.
 | ||
| Fig. 10 Mechanism diagram of the RhB photodegradation on PbBiO2Br/UiO-66-NH2. | ||
4. Conclusions
In summary, PbBiO2Br/UiO-66-NH2 heterojunctions have been successfully prepared by a facile, one-pot solvothermal method. They have been investigated for the photodegradation of RhB dye solution and colorless phenol solution under visible light irradiation, and for two model organic pollutants, the heterojunctions all exhibits better photocatalytic activity than pure PbBiO2Br and UiO-66-NH2. For RhB degradation, the PbBiO2Br/UiO-66-NH2 (3:2) sample displays the highest degradation efficiency, the reaction rate of which is about 5 and 4 times as fast as that of PbBiO2Br and UiO-66-NH2, respectively. In addition, photogenerated holes and O2˙− radicals are believed to be the main active species responsible for photocatalysis.
Acknowledgements
This work was financially supported by the Anhui Provincial Natural Science Foundation (No. 1508085MB28) and the National Natural Science Foundation of China (Grant. 51372062).References
- A. S. Bhatt, P. L. Sakaria, M. Vasudevan, R. R. Pawar, N. Sudheesh, H. C. Bajaj and M. Mody, RSC Adv., 2012, 2, 8663–8671 RSC.
- H. Chen and J. Zhao, Adsorption, 2009, 15, 381–389 CrossRef CAS.
- V. K. Gupta, B. Gupta, A. Rastogi, S. Agarwal and A. Nayak, J. Hazard. Mater., 2011, 186, 891–901 CrossRef CAS PubMed.
- A. Dolbecq, P. Mialane, B. Keita and L. Nadjo, J. Mater. Chem., 2012, 22, 24509–24521 RSC.
- A. Kubacka, M. Fernández-García and G. Colón, Chem. Rev., 2012, 112, 1555–1614 CrossRef CAS PubMed.
- W. Q. Fan, Q. H. Zhang and Y. Wang, Phys. Chem. Chem. Phys., 2013, 15, 2632–2649 RSC.
- Z. Yi, J. Ye, N. Kikugawa, T. Kako, S. Ouyang, H. Stuart-Williams, H. Yang, J. Cao, W. Luo, Z. Li, Y. Liu and R. L. Withers, Nat. Mater., 2010, 9, 559–564 CrossRef CAS PubMed.
- L. Wu, H. B. Jiang, F. Tian, Z. Chen, C. Sun and H. G. Yang, Chem. Commun., 2013, 49, 2016–2018 RSC.
- J. Jiang, K. Zhao, X. Xiao and L. Zhang, J. Am. Chem. Soc., 2012, 134, 4473–4476 CrossRef CAS PubMed.
- J. Ungelenk and C. Feldmann, Chem. Commun., 2012, 48, 7838–7840 RSC.
- N. Zhang, S. Ouyang, T. Kako and J. Ye, Chem. Commun., 2012, 48, 9894–9896 RSC.
- D. O. Charkin, P. S. Berdonosov, V. A. Dolgikh and P. Lightfoot, J. Solid State Chem., 2003, 175, 316–321 CrossRef CAS.
- M. Cherevatskaya, M. Neumann, S. Füldner, C. Harlander, S. Kümmel, S. Dankesreiter, A. Pfitzner, K. Zeitler and B. König, Angew. Chem., Int. Ed., 2012, 51, 4062–4066 CrossRef CAS PubMed.
- Z. C. Shan, W. D. Wang, X. P. Lin, H. M. Ding and F. Q. Huang, J. Solid State Chem., 2013, 3, 10687–10690 Search PubMed.
- G. Q. Song, Z. Q. Wang, L. A. Wang, G. R. Li, M. J. Huang and F. X. Yin, Chin. J. Catal., 2008, 181, 1361–1366 CrossRef.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- H. L. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS.
- P. Wang, J. Feng, Y. P. Zhao, S. B. Wang and J. Liu, ACS Appl. Mater. Interfaces, 2016, 8, 23755–23762 CAS.
- S. Kitagawa, R. Kitaura and S. I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- S. Hasegawa, S. Horike, R. Matsuda, S. Furukawa, K. Mochizuki, Y. Kinoshita and S. Kitagawa, J. Am. Chem. Soc., 2007, 129, 2607–2614 CrossRef CAS PubMed.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS PubMed.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. OõKeeffe and O. M. Yaghi, Science, 2002, 295, 469–472 CrossRef CAS PubMed.
- M. Latroche, S. Surblé, C. Serre, C. Mellot-Draznieks, P. L. Llewellyn, J. H. Lee, J. S. Chang, S. H. Jhung and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 8227–8231 CrossRef CAS PubMed.
- P. Horcajada, C. Serre, M. Vallet-Regí, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., 2006, 118, 6120–6124 CrossRef.
- C. G. Silva, I. Luz, F. X. Llabrés i Xamena, A. Corma and H. García, Chem.–Eur. J., 2010, 16, 11133–11138 CrossRef CAS PubMed.
- T. W. Goh, C. X. Xiao, R. V. Maligal-Ganesh, X. L. Li and W. Y. Huang, Chem. Eng. Sci., 2015, 124, 45–51 CrossRef CAS.
- L. Shen, W. Wu, R. Liang, R. Lin and L. Wu, Nanoscale, 2013, 19, 9374–9382 RSC.
- M. A. Aramendía, V. Borau, J. C. Colmenares, A. Marinas, J. M. Marinas, J. A. Navío and F. J. Urbano, Appl. Catal., B, 2008, 80, 88–97 CrossRef.
- M. Zhou, J. Yu, S. Liu, P. Zhai and L. Jiang, J. Hazard. Mater., 2008, 154, 1141–1148 CrossRef CAS PubMed.
- F. Y. Xiao, J. Xing Long, W. Z. P. Chen, X. L. Wang and H. G. Yang, RSC Adv., 2013, 3, 10687–10690 RSC.
- X. Zhang, F. Jia and L. Zhang, J. Phys. Chem. C, 2008, 112, 747–753 CAS.
- X. F. Cao, L. Zhang, X. T. Chen and Z. L. Xue, CrystEngComm, 2011, 13, 306–311 RSC.
- M. A. Butler, J. Appl. Phys., 1914, 48, 1914–1920 CrossRef.
- Q. Chen, Q. Q. He, M. M. Lv, Y. L. Xu, H. B. Yang, X. T. Liu and F. Y. Wei, Appl. Surf. Sci., 2015, 327, 77–85 CrossRef CAS.
- D. Chen, Z. Jiang, J. Geng, Q. Wang and D. Yang, Ind. Eng. Chem. Res., 2007, 46, 2741–2746 CrossRef CAS.
- L. Mohapatra, K. Parida and M. Satpathy, J. Phys. Chem. C, 2012, 116, 13063–13070 CAS.
- P. Xiong, Q. Chen, M. Y. He, X. Q. Sun and X. Wang, J. Mater. Chem., 2012, 22, 17485–17493 RSC.
- P. Xiong, L. J. Wan, X. Q. Sun, B. H. Xu and X. Wang, Ind. Eng. Chem. Res., 2013, 53, 10105–10113 CrossRef.
- Q. Chen, Q. Q. He, M. M. Lv, X. T. Liu, J. Wang and J. P. Lv, Appl. Surf. Sci., 2014, 311, 230–238 CrossRef CAS.
- X. Wang, S. O. Pehkonen and A. K. Ray, Ind. Eng. Chem. Res., 2004, 43, 1665–1672 CrossRef CAS.
- A. J. Bard and L. R. Faulkner, Electrochemical methods fundamentals and applications, John Wiley & Sons, New York, 1980 Search PubMed.
- T. X. Wu, G. M. Liu, J. C. Zhao, H. Hidaka and N. Serpone, J. Phys. Chem. B, 1998, 102, 5845–5851 CrossRef CAS.
- Q. Chen, Q. Q. He, M. M. Lv, Y. L. Xu, H. B. Yang, X. T. Liu and F. Y. Wei, Appl. Surf. Sci., 2015, 327, 77–85 CrossRef CAS.
- J. M. Liu, Q. C. Zhang, J. C. Yang, H. Y. Ma, M. O. Tade, S. B. Wang and J. Liu, Chem. Commun., 2014, 50, 13971–13974 RSC.
| This journal is © The Royal Society of Chemistry 2016 |