
Toward synergy of carbon and La2O3 in their hybrid as an efficient catalyst for the oxygen reduction reaction
Nan Wang,
Jingjun Liu*,
Weiwei Gu,
Ye Song and
Feng Wang*
State Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology for Materials, Beijing University of Chemical Technology, Beijing 100029, PR China. E-mail: liujingjun@mail.buct.edu.cn; wangf@mail.buct.edu.cn; Fax: +86-10-64411301; Fax: +86-10-64451996; Tel: +86-10-64411301 Tel: +86-10-64451996
First published on 9th August 2016
Abstract
Investigating the synergy in hybrids between rare earth (La, Ce, Y) oxides and carbon may be an effective way to develop new efficient and cheap catalysts for catalyzing the oxygen reduction reaction (ORR). In this work, a set of La2O3/C hybrids with different La2O3 loadings were prepared by chemical precipitation in alkaline solution, followed by calcination treatment. The prepared La2O3 nanoparticles with hexagonal structure covered uniformly on the carbon surface. X-ray photoelectron spectroscopy (XPS) indicated different contents of covalent C–O–La bonds at the interface between the La2O3 and carbon in these hybrids. The electrochemical experiments in alkaline solution show that the catalyst with 80 wt% of La2O3 exhibits the highest electrochemical activity in catalyzing ORR and the lowest production of hydrogen peroxide among the synthesized hybrids. The remarkably enhanced ORR activity is attributed to the maximum content of the C–O–La bonds formed in the hybrid. Interestingly, the above C–O–La covalent bonds can promote the electron transfer from the supported carbon (π electron) to the La2O3 phase, and the transferred electron can fill the unoccupied eg orbital splitted by the La 5d orbital of La2O3, which should be responsible for the improved ORR performance.
1. Introduction
The oxygen reduction reaction (ORR) has received more and more attention due to its extensively practical applications in energy storage and conversion devices.1–6 However, due to the sluggish kinetic process of the ORR in acidic or alkalline environments,7 electro-catalysts are essentially required to achieve efficient utilization of the ORR in these above fields. Currently, considered as the best catalyst for the ORR, the Pt/C catalyst has superior catalytic activity, but its high cost, scarcity and lower stability limit its practical utility.8The rare earth oxides, such as La2O3, CeO2, Y2O3, etc., as functional materials, have been extensively investigated for their excellent electronic, photo-catalytic and electro-catalytic properties arised from their unique electronic structures.9 The advantages of these rare earth oxides as catalysts can be obtained from the following two respects. On the one hand, lanthanides, such as La and Ce, are located in f-block and have characteristic 4f electron configuration, which can contribute the migration of 4f electrons between f–f configuration and f–d configuration to form unique bonding characteristics. These bonding characteristics would reduce the activation energy and influence the process of catalyzing the ORR.10–12 On the other hand, lanthanum oxides contain oxygen vacancies and interstitials with low oxygen vacancy energy, which makes the oxygen vacancies readily supplemented by rapid oxygen diffusion through the oxide and to the surface, leading to low activation energy.13,14 Furthermore, the existed inter-layer defect structure of the oxides is also helpful to the active oxygen adsorption, which may have a positive effect on catalyzing the ORR. Although the lanthanum oxides have outstanding electronic structure, La2O3 is not electroconductive, which limits its exertion in electro-catalysis.
Nowadays, La2O3 supported on a carbon (carbonaceous micro-sphere) (La2O3@CMSs) has been synthesized by a hydrothermal treatment of glucose precursor and exhibits high ORR catalytic activity in alkaline solution.15 And, the improved performance would be ascribed to the formation of the active components of La–O and C–O bonds at the interface between the carbon and La2O3 phase in this hybrid. The effect of the interfacial interaction between carbon and La2O3 phase on the ORR activity has been confirmed by Liu et al.16 In this work, La2O3 supported on a carbon black has been fabricated and shows surprisingly high catalytic activity and stability toward the ORR with respect to the pure oxide or carbon alone. It is believed that the covalent C–O–La bonds, formed at the interface between lanthanum oxide and carbon components, are responsible for the enhanced electro-catalytic performance toward the ORR. Hence, the strong chemical interaction between carbon and La2O3 phase may play a key role in improving the ORR activity. The detail reasons for the improved activity can be explained by a strong electron transfer from the supported carbon to the metallic oxide, raised by the above chemical covalent bonds. This finding has been verified by recently published papers.17,18 The possible interfacial interaction between the carbon materials and rare earth oxides has been also confirmed by some investigators.19,20 Hu et al.19 synthesized the silver modified LaMnO3-graphene catalyst, which shows excellent catalytic activity to the ORR. In the work, such enhanced electro-catalytic activity was attributed to the synergistic effect of LaMnO3 and graphene in the composite. Liu et al.20 prepared modified carbon black-LaMnO3 catalysts with improved electro-catalytic activity to the ORR and they confirmed that the formation of covalent bonds C–O–M (M = Mn, La) between the LaMnO3 and carbon would effectively enhance the ORR kinetics. Besides, Sachin Kumar et al.21 analyzed electron transfer and synergistic effect, which enhanced the photo-catalytic activity of rGO–CeO2 nanocomposites, between reduced graphene oxide (rGO) sheets and CeO2 nanoparticles. Hence, we can conclude that the strong covalent electron transfer between rare-earth oxides and supported carbon materials, can favour the ORR activity. In this way, the controlling of the covalent electron transfer may be a feasible and effective means to develop electro-catalytic materials, because such electron transfer process can efficiently modulate the electronic structures of active atoms, such as La3+ 5d and/or O 2p in La2O3, which may play a key role in lowering the activation energy of the ORR in nature. However, the detail process of the electron transfer remains unclear, since the change in the electron configurations of La2O3 phase on carbon has been paid little attention, which hinders the development of hybrids as efficient catalysts for ORR.
In this work, we proposed a simple method to synthesize La2O3 using carbon black as the support, which is an efficient electro-catalyst for ORR, via a urea precipitation method. We characterized the electronic structures of these hybrids by XPS measurements. And then, the electro-catalytic activities of the synthesized hybrids depended on their composition toward the ORR in alkaline solution were measured. More importantly, it is detected profoundly that effects of the covalent electron, transferred from the carbon to the La2O3, on the electronic structures of La2O3 or carbon components in the prepared hybrid. The change in La3+ 5d and/or O 2p electron configurations in La2O3 probably contributes to the activity. This explored information is essential for profound understanding the physical origin of the electro-catalytic performance of La2O3/C catalysts toward the ORR.
2. Experimental
2.1 Preparation of La2O3/C hybrids
A set of La2O3/C hybrids with different La2O3 loadings (40 wt%, 60 wt%, 80 wt% and 100 wt%, relative to carbon) were prepared by a chemical precipitation method in alkaline solution system. For all the synthesized hybrids, all reagents are analytic grade and purchased from Sinopharm Chemical Co., Ltd. Prior to lanthanum oxide deposition, pretreating carbon black (Vulcan XC-72) with concentrated nitrate acid (HNO3, 14.6 M). Then, the acid-treated carbon black was placed in a vacuum to dry after being filtered and washed by deionized water. Afterwards, lanthanum oxide was deposited on the surface of the acid-oxidized carbon by a chemical precipitation of LaCl3 on the carbon black in alkaline aqueous solution, using urea as a precipitating agent. Typically, a certain amount of the treated carbon was dispersed in the mixture of 20 mL of 0.05 M lanthanum chloride (LaCl3) and 0.25 wt% of ammonium sulphate ((NH4)2SO4) to obtain a black suspension. Subsequently, 20 mL of 0.15 M urea and 24.8 mM of cetyl trimethyl ammonium bromide (CTAB) were added to the above black suspension. The obtained mixture was aged for 2 h for sufficient reaction, and then ultrasonicated for 40 min. After that, filtering and washing the reaction mixture with deionized water and anhydrous ethanol, until no chlorine ion in the solution, then drying the reaction mixture in an oven at 80 °C for 2 h. Finally, annealing the above mixture in tube furnace under argon flow at 750 °C for 2 h to produce the lanthanum oxide on carbon.2.2 Physical characterizations
X-ray diffraction (XRD) experiments for these La2O3–carbon hybrids were measured with Rigaku RINT 2200 V/PC using Cu Kα radiation (λ = 1.5406 Å) with a scanning rate of 5° min−1. Scanning electron microscopy (SEM) (FE-SEM, JEOL, JSM-6701F) and transmission electron microscopy (TEM) were used to analyze morphological features of these hybrids. X-ray photoelectron spectroscopy (XPS, Thermo ESCALAB 250) was operated to explore electronic structures of these samples. Herein, the performed XPS spectra were calibrated by the C 1s spectrum with the binding energy at 285.0 eV. Raman spectra were performed to analyze the electron transfer in these as-prepared hybrids from the carbon to the lanthanum oxide.2.3 Electrochemical measurements
We studied the ORR catalytic activity of these La2O3/C catalysts with rotating ring-disk electrode (RRDE) in alkaline solution with saturated oxygen. In the measured system, we use the saturated calomel electrode (SCE) as the reference electrode, Pt foil as the counter electrode and the glassy carbon electrode (S = 0.247 cm2) pipetted with the catalyst suspension as the working electrode, respectively. During the process of the electrochemical measurements, we should keep the saturation of oxygen gas in the testing solution. For all the electrochemical data given in the work, potentials were transformed to the reversible hydrogen electrode (RHE) by calibrating with the formula Evs. RHE = Evs. SCE + 0.241 V + 0.059 pH.22,23 Moreover, chronoamperometric response mearsurements for the hybrids were carried out with electrochemical workstation system (CHI 600E) in 1 M NaOH for 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 s.
000 s.
3. Results and discussion
3.1 Fabrication and formation mechanism for La2O3/C hybrid
Fig. 1 shows the morphologies of the synthesized La2O3/C hybrids with different La2O3 loadings. As shown in Fig. 1(A)–(D), the La2O3 nanoparticles deposited evenly on the surface of the carbon with similar particle sizes, without apparent agglomeration. As observed from the SEM images in Fig. 1(A)–(D), these nanoparticles have a size of about 10–20 nm but slightly grow up as the La2O3 loadings increase from 40 wt% to 100 wt%. The slight agglomeration of La2O3 nanoparticles for 100 wt% of La2O3/C hybrid is reasonably attributed to the insufficient functional groups such as carboxyl and hydroxyl on carbon surface as La2O3 loadings increase. Besides, according to these TEM images, the observed lattice fringes, composed by very straight lines, should result from the oxide rather than the carbon support, because the lattice fringes of the carbon black are generally of wrinkles. So, the lattice distances in the La2O3 particles are all about 3.4 Å and conform to the (100) crystal plane of hexagonal La2O3 nanocrystals, which reveals the presence of hexagonal La2O3 phase. | ||
| Fig. 1 SEM images and TEM images (inset) of La2O3/C hybrids prepared at different La2O3 loadings of (A) 40 wt%, (B) 60 wt%, (C) 80 wt% and (D) 100 wt%; (E) XRD patterns of La2O3/C hybrids above; (F) schematic structure of La2O3 nanoparticles with hexagonal phase. | ||
To further analyze the lattice structure of these La2O3/C hybrids, XRD patterns were performed, and the obtained results are displayed in Fig. 1(E). Apart from the diffraction peak at about 26° originated from the face-to-face stacking of the (002) crystalline plane of carbon, the eight diffraction peaks, located at 26.1°, 29.9°, 39.5°, 46.1°, 52.1°, 55.4°, 72.1° and 75.3° respectively, can be indexed to the (100), (101), (102), (110), (103), (112), (203) and (211) planes of hexagonal La2O3 (JCPDF-05-0602). The XRD patterns further confirm that these La2O3 nanoparticles have pure hexagonal phase structure in the hybrids without impurities since no additional peaks displayed. The lattice structure of the synthesized hexagonal-phase La2O3 can be described by Fig. 1(F). As observed, the La2O3 phase belongs to the A-type structure of rare-earth oxides among three types of structure with A-, B-, and C-type.24 In this A-type structure, the oxygen form hexagonal close packed (hcp) unit cells, while the large-size lanthanum cations locate in the center of the octahedral interstices formed by six oxygen anions, since the octahedral interstice is larger than the tetrahedral interstices.25 A symmetrical stacking of La atoms in O–La–O–La–O structures is presented in the La2O3 phase, as suggested by the researchers.24 In addition, based on XRD patterns in Fig. 1(E), the average sizes of La2O3 nanoparticles are measured with Scherrer's equation26 and range from 9.84, 10.01, 12.28 and 13.84 nm for the hybrids with a loading range from 40 wt% to 100 wt%.
The formation mechanism for the La2O3/C hybrids involves a continual process of nucleation and growth of the lanthanum oxide supported on the carbon surface. Since CTAB as surfactant is a special hydrophilic/hydrophobic compound including a hydrophobic long-chain (tail) and a hydrophilic polar group (head) in its structure, this surfactant will tend to self-associate into micellars in aqueous solutions, with double-layer hydrophilic/hydrophobic structure that is favored thermo-dynamically in aqueous solutions.16,27,28
These micelles in aqueous solutions may be responsible for the space-confined growth of the oxide nanoparticles, at a critical micelle concentration (CMC) of CTAB. The detail growth mechanism of La2O3 on carbon can be explained as shown in Fig. 2. As seen in Fig. 2(A), these small micelles formed by CTAB can wrap the La3+ ions and urea, where the factant surface layer formed by CTAB, as space barrier, can control mixture of two reactants (LaCl3 and urea) and overgrowth of La2O3 nanoparticles on carbon.29 Subsequently, the space-confined La3+ ions and urea react each other to form La(OH)CO3 nucleations, as shown in Fig. 2(B). These La(OH)CO3 nucleations can grow by consumption of the La3+ and urea to form small La(OH)CO3 domains on carbon in these micelles, as shown in Fig. 2(C). Then the obtained La(OH)CO3 domains are calcined at 750 °C under the argon flow, during which (NH4)2SO4 decomposes into N2, SO2 and NH3 to prevent the particle agglomeration. Eventually, La2O3 nanoparticles are formed on the carbon with uniform dispersion as shown in Fig. 2(D).
 | ||
| Fig. 2 Schematic formation mechanism of pure hexagonal La2O3 nanoparticles supported on carbon black. (A) The formation of micelles by CTAB on carbon; (B) the formation of La(OH)CO3 nucleations on carbon; (C) the formation of La(OH)CO3 precursor domains on carbon; (D) the formation of La2O3 oxides on carbon. | ||
3.2 Electro-catalytic performance of La2O3/C hybrids for ORR
To study electro-catalytic activity of the synthesized La2O3/C hybrids for ORR, a set of the rotating disk polarization curves were recorded in 1 M NaOH solution with saturated oxygen, as shown in Fig. 3(A). Considering onset potential can reflect the trend of ORR activity, we determined the onset potentials of these hybrids using the data in Fig. 3(A) to evaluate their activities and displayed in Fig. 3(B). As observed, the onset potentials of these La2O3/C hybrids show the order with 80 wt% > 100 wt% > 60 wt% > 40 wt%, with the values of 0.90 V, 0.86 V, 0.84 V and 0.81 V (vs. RHE), respectively. It is also noticed that the 80 wt% La2O3/C catalyst has the best catalytic activity among all the tested catalysts, which exceeds that obtained from the other tested hybrid catalysts. As evidenced in Fig. 3(C), based on the data at a potential of 0.80 V (vs. RHE), the kinetic current density (JK) of oxygen reduction is 4.09 mA cm−2 for the catalyst, which is much higher than that obtained from the other tested hybrid catalysts. The kinetic current density (JK) can be calculated by the following equation (eqn (1)):
 | (1) |
 | ||
| Fig. 3 (A) Polarization curves for La2O3/C hybrids with different La2O3 loadings recorded at a fixed rotation rate of 1600 rpm and scan rate of 5 mV s−1 in an O2-saturated 1 M NaOH solution at room temperature; (B) onset potential of these hybrids; (C) the kinetic current density of these hybrids at a given potential of 0.80 V (vs. RHE); (D) mass specific activity of La2O3/C hybrids. | ||
To gain some insight to the effect of the La2O3 phase on the ORR activity, we also plotted the mass-specific activity of these La2O3/C catalysts as a function of the La2O3 loadings using the data in Fig. 3(C). As depicted in Fig. 3(D), a similar volcano-trend along with La2O3 loadings has been observed for these La2O3/C catalysts, where the 80 wt% catalyst locates at the peak. At the potential of 0.80 V (vs. RHE), the mass specific activity of the catalyst is 23.86 mA mg−1, which is the highest among all the tested catalysts. This outcome reveals that the improved activity of the La2O3/C catalysts in catalyzing ORR would be attributed to the strong interfacial interaction between the La2O3 and supported carbon at their interfaces, although the contribution of the oxide to the electro-catalysis should be not neglected.16–18
To investigate the pathway of the ORR on the La2O3/C catalysts, the H2O2 yields on them were performed using rotating ring-disk measurements in 1 M NaOH solution with saturated O2.
The following equation (eqn (2)) was used to calculate hydrogen peroxide productions and the obtained results are shown in Fig. 4(A).30 As observed, the determined hydrogen peroxide production on the catalyst (80 wt% La2O3) is much lower than that on the other tested catalysts. The lower H2O2 yield reveals a near four-electron pathway through the ORR electro-catalytic process for the catalyst. To further prove the pathway of the ORR catalyzed by the La2O3/C catalyst, the rotating disk electrode measurements were performed at different rotation speeds for the sample, as shown in Fig. 4(B). Moreover, Fig. 4(C) shows the number of electrons transferred per O2 molecule (n) for all the tested catalysts with different La2O3 loadings, calculated from the slope of Koutecky–Levich plots (J−1 versus ω−1/2) (eqn (3)). As observed, the La2O3/C hybrid fabricated at 80 wt% La2O3 shows the highest electron transfer number for the ORR among these tested catalysts, illustrating the ORR catalyzed by the catalyst proceeds nearly through the four-electron pathway. It is consistent with the results in Fig. 4(A).
 | (2) |
 | (3) |
485 C mol−1), C0 is the bulk concentration of O2 in 1 M NaOH (0.843 × 10−6 mol cm−3), D0 is the diffusion coefficient of oxygen in 1 M NaOH (1.43 × 10−5 cm2 S−1), v is the kinematic viscosity of the electrolyte (0.01128 cm2 S−1), and ω is the angular velocity.
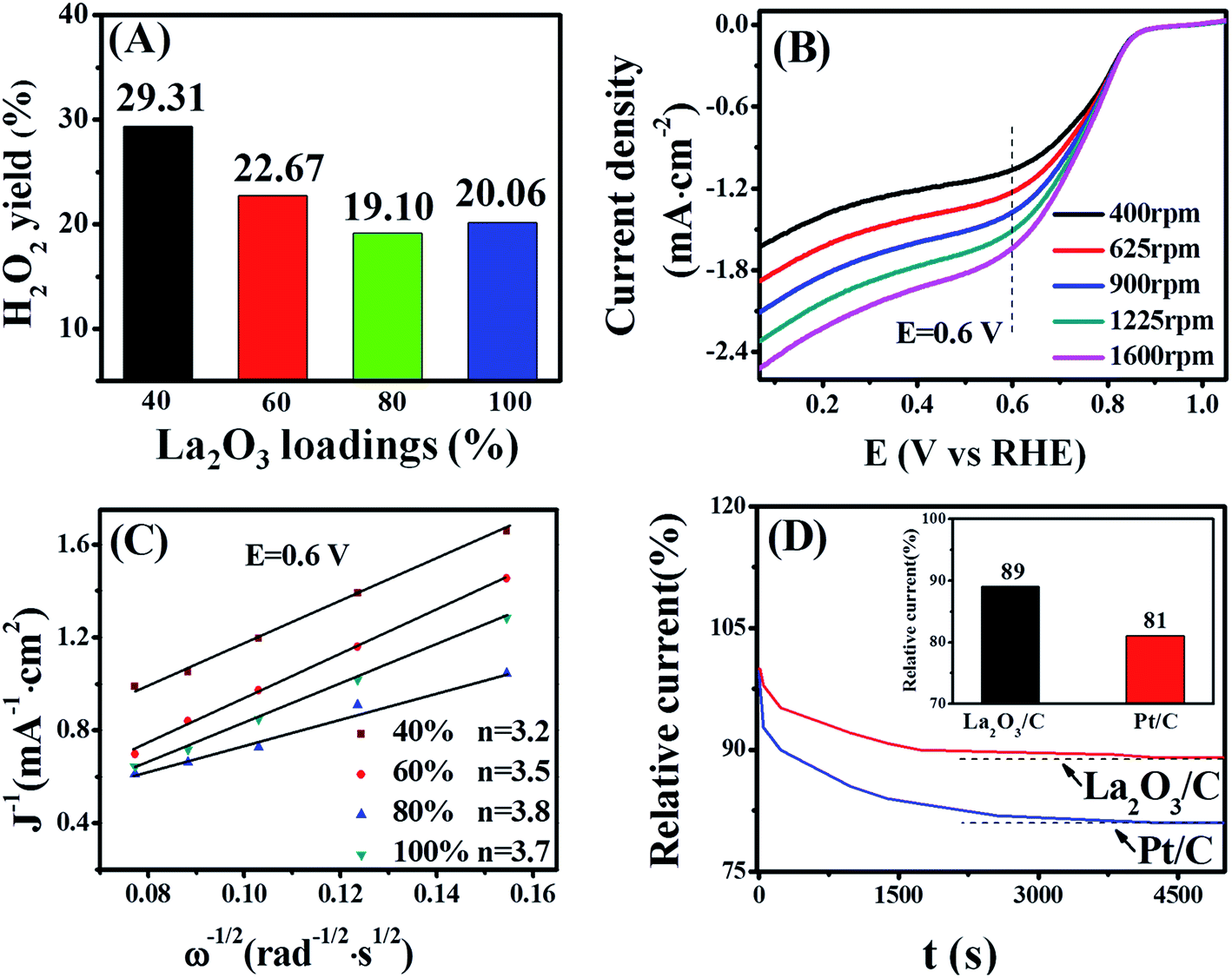 | ||
| Fig. 4 (A) Hydrogen peroxide yields on the synthesized La2O3/C hybrid catalysts at a given potential of 0.60 V (vs. RHE); (B) rotating disk electrode voltammograms of the 80 wt% La2O3/C sample at various rotation speeds at scan rate of 5 mV s−1; (C) Koutecky–Levich plots (J−1 versus ω−1/2) for ORR on La2O3/C hybrids with different La2O3 loadings obtained at 0.60 V (vs. RHE); (D) chronoamperometric responses of the hybrid and the commercial Pt/C (E-TEK) during the ORR in 1 M O2-saturated NaOH solution at 0.60 V(vs. RHE) at 900 rpm. | ||
Moreover, the long-term stability of the catalyst was analyzed by chronoamperometric curves in 1 M NaOH solution with saturated O2, as shown in Fig. 4(D). The oxygen reduction current for the catalyst is slightly decline to 89% relative to that of a commercial Pt/C (20 wt%, E-TEK) for 81%, revealing the ORR performance of La2O3/C catalyst is much more stable than that of the commercial Pt/C.
3.3 Chemical interaction between oxide and carbon
To identify chemical interaction between the La2O3 and carbon in their hybrid, X-ray photoelectron spectroscopy (XPS) were performed for the synthesized La2O3/C hybrids with different La2O3 loadings, as shown in Fig. 5. The fitted C 1s spectra for these samples are shown in Fig. 5(A). It displays three characteristic peaks at about 285.0 eV, 286.7 eV and 289.0 eV, which corresponds to C–C, C–O and O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds in the acid-treated carbon support, respectively.31 Besides, these La2O3/C hybrids also exhibit a new peak at 286.2 eV in Fig. 5(A). It may be related to the covalent C–O–La bonds arised from the chemical interaction between La2O3 and carbon in their catalyst, according to the literature.16,20 To confirm this conjecture, the O 1s spectra have been fitted for these samples too. As shown in Fig. 5(B), these La2O3/C hybrids show four characteristic peaks at around 531.7 eV, 532.1 eV, 532.9 eV and 533.7 eV, respectively. The two peaks among them at about 533.7 eV and 532.1 eV should be attributed to C–O and O–CO component existed on the carbon surface.31 Another observed peak at near 531.7 eV should be ascribed to the La–O bonds in La2O3 phase.15 And the last peak at about 532.9 eV would be arised from C–O–La covalent bonds, in which the carbon atoms connected with lanthanum atoms through oxygen atoms, as suggested in the literature.17,20 It reveals the presence of the chemical interaction between the carbon and La2O3 in their hybrids.
O bonds in the acid-treated carbon support, respectively.31 Besides, these La2O3/C hybrids also exhibit a new peak at 286.2 eV in Fig. 5(A). It may be related to the covalent C–O–La bonds arised from the chemical interaction between La2O3 and carbon in their catalyst, according to the literature.16,20 To confirm this conjecture, the O 1s spectra have been fitted for these samples too. As shown in Fig. 5(B), these La2O3/C hybrids show four characteristic peaks at around 531.7 eV, 532.1 eV, 532.9 eV and 533.7 eV, respectively. The two peaks among them at about 533.7 eV and 532.1 eV should be attributed to C–O and O–CO component existed on the carbon surface.31 Another observed peak at near 531.7 eV should be ascribed to the La–O bonds in La2O3 phase.15 And the last peak at about 532.9 eV would be arised from C–O–La covalent bonds, in which the carbon atoms connected with lanthanum atoms through oxygen atoms, as suggested in the literature.17,20 It reveals the presence of the chemical interaction between the carbon and La2O3 in their hybrids.
 | ||
| Fig. 5 XPS spectra of La2O3/C hybrids with different La2O3 loadings (40 wt%, 60 wt%, 80 wt% and 100 wt%): (A) C 1s (B) O 1s and (C) La 3d 5/2; (D) the content of C–O–La bonds within these of La2O3/C hybrids above. | ||
To further evidence the chemical interaction, we also fitted the La 3d spectra of these samples, as shown in the Fig. 5(C). As observed, the La 3d5/2 core levels splitted into the basic peaks (about 836.1 eV) and the separated satellite peak at a higher binding energy (about 840.0 eV).32 The satellite peak shifted positively along with the increasing loading of the La2O3 but the 80 wt% La2O3/C shows the highest binding energy of the satellite peak among all the samples. The strong shift in binding energy would be assigned to the strong interfacial chemical interaction between La2O3 and carbon in the hybrids, which is coinciding with the analysis in Fig. 5(A) and (B).
Except for the unique chemical interaction between the La2O3 and carbon shown in Fig. 5(A)–(C), we also noticed a strong relationship between the La2O3 loadings and contents of the covalent C–O–La bonds formed in these hybrids. As observed in Fig. 5(A) and (B), it is obvious that the intensity of the characteristic peak, assigned to the C–O–La bonds, is different for these La2O3/C hybrids with different loadings. Fig. 5(D) shows the detailed relationship between the La2O3 loadings and the contents of the C–O–La bonds that are calculated by the peak areas referring to the above C–O–La bonds in the C 1s spectra. As observed, the contents of the peak increase firstly and then decrease as a function of the oxide loadings from 40 wt% to 100 wt%; the hybrid with 80 wt% loading has the largest content of the covalent C–O–La bond among all the tested hybrids. In comparison with the 80% La2O3/C catalyst, however, the 100% catalyst displays a slightly decrease in the bond content, which is due to the slight agglomeration of La2O3 particles on carbon, as shown in Fig. 1(D). The tendency matches well the content variation for the same peak, recorded in O 1s spectra shown in Fig. 5(B). Moreover, the shift of La 3d spectra shown in Fig. 5(C) also confirms this result. So, these findings provide a direct proof for the presence of the close correlation between the oxide loadings and the contents of the C–O–La bonds formed in these hybrids.
As suggested by some published works,18,31,33 for the oxide-carbon hybrids, the C–O–La bonds formed at the interface may be responsible for the enhanced ORR activity of the La2O3/C catalysts. So, we plotted the JK of the La2O3/C catalysts shown in Fig. 3(C) as a function of the contents of the C–O–La bonds using the data in Fig. 4(D). The obtained results are shown in Fig. 6(A). As expected, as the content of the C–O–La bonds increases, the JK increases correspondingly, which exhibits an almost linear trend, indicating a close correlation between ORR catalytic activity and the content of the bond. Moreover, the mass specific activity of each sample was also plotted along with its content of the C–O–La bonds, as shown in Fig. 6(B). The similar tendency has been found in Fig. 6(A) and (B). Taken together, these results suggest that the increased content of the C–O–La bonds at the interface in these hybrids facilitates the ORR activity of the La2O3/C hybrid catalysts. As shown in Fig. 3, the catalyst with 80 wt% La2O3 exhibits the highest ORR activity among all the tested catalysts, because of its highest content of the bond. However, as the loading of La2O3 increased to 100 wt%, owing to the slight agglomeration of the nanoparticles shown in Fig. 1(D), the content of the C–O–La bonds correspondingly decrease slightly for the 100 wt% La2O3/C catalyst shown in Fig. 5(D), in comparison with the 80% La2O3/C. More importantly, at this case, the reduced electrical conductivity for the hybrid (100 wt% La2O3/C) would also decay the catalytic activity toward the ORR. Therefore, the improved ORR activity of the La2O3/C catalysts, indeed, originates from the strong chemical interaction between the oxide and carbon at the interface in their hybrids, as shown in Fig. 6(C). Therefore, to enhance synergistic interaction between lanthanum oxide and carbon components may be a simple and feasible way to fabricate new and excellent ORR catalysts.
 | ||
| Fig. 6 (A) Relationship between kinetic current density (JK) and contents of the covalent C–O–La bonds; (B) relationship between mass specific activity and contents of the covalent C–O–La bonds; (C) schematic illustration of the unique interfacial structure between La2O3 and carbon in the hybrids and the overall ORR process catalyzed by the La2O3/C hybrids. | ||
3.4 Origin of electro-catalytic activity for La2O3/C hybrids
To elaborate deeply the physical origin of the activity of the La2O3/C catalyst for ORR arised from the covalent C–O–La bonds at their interface, we performed a rotating ring-disk electrode (RRDE) measurement for the La2O3/C catalyst (80 wt% La2O3/C), La2O3 physically mixed with carbon (La2O3 + C), and a commercial Pt/C catalyst with 20% Pt (E-TEK) as reference. Concerning the RRDE results shown in Fig. 7(A), the onset potential and kinetic current for each catalyst have been extracted, respectively. As observed in Fig. 7(B), the onset potential toward the ORR is negatively shifted by ∼50 mV for the covalent La2O3/C hybrid relative to the commercial Pt/C but far exceeding that obtained from the physical mixture sample (La2O3 + C). The trend of the onset potentials matches well the kinetic current density (JK) for the ORR (Fig. 7(C)). As evidenced from Fig. 7(D), the mass-specific activity of La2O3/C composite is lower than that obtained from the commercial Pt/C, but much higher than that obtained from the La2O3 physically mixed with carbon. The mass-specific activity of the La2O3/C catalyst is 3.84 times higher than that of the physical mixture. At the same time, the hydrogen peroxide yield of the La2O3/C catalyst is higher than that obtained from the commercial Pt/C, but lower than that obtained from the physical mixture of La2O3 and carbon, as shown in Fig. 7(E). With respect to the mixture catalyst, the lower hydrogen peroxide production on the La2O3/C catalyst is attributed to the formation of the covalent C–O–La bonds at the interface between the La2O3 and carbon, as shown in Fig. 5 and 6. The impact of the covalent C–O–La bonds on the hydrogen peroxide yield is shown in Fig. 4. As observed, the chemical interaction between oxide and carbon can facilitate the ORR following a nearly four-electron pathway catalyzed by the hybrid. This finding is in agreement with the results obtained by recently published papers.16–18 Therefore, these outcomes indeed confirm that the remarkably enhanced electro-catalytic activity of the hybrid is mainly attributed to the unique chemical interaction between La2O3 and carbon arised from the covalent C–O–La bonds formed at the interface in their hybrid. | ||
| Fig. 7 (A) Polarization curves for blend sample, 80 wt% La2O3/C hybrid and commercial Pt/C catalyst recorded at a fixed rotation rate of 1600 rpm and scan rate of 5 mV s−1 in 1 M O2-saturated NaOH solution at room temperature; (B) onset potential of these samples; (C) the kinetic current density of these samples at a given potential of 0.80 V (vs. RHE); (D) mass specific activity of these samples at a given potential of 0.80 V (vs. RHE); (E) hydrogen peroxide yields on these samples at a given potential of 0.60 V (vs. RHE). | ||
According to recently published results,16 the chemical interaction can promote the electron transfer from the carbon to La2O3 phase by the C–O–La bonds at the interface, which can facilitate the ORR kinetics. So, in our case, Raman spectra were measured for the La2O3/C catalyst to explore the electron transfer process in it, as shown in Fig. 8(A). Two regular peaks of carbon, which are assigned to D peak at 1348 cm−1 and G peak at 1584 cm−1, are displayed on the performed Raman spectra. However, a slightly positive shift of the G peak was observed for the hybrid with 1603 cm−1 compared with 1584 cm−1 of the pure carbon, which reveals the electron transfer from the carbon to La2O3.16,31,34 Besides, Raman spectra for the La2O3 phase in the hybrid and pure La2O3 were performed respectively, as shown in Fig. 8(B), which may give more information about the unique electron transfer process. Two characteristic peaks of pure La2O3 have been observed in Fig. 8(B). The one of them is Eg peak at 408 cm−1 ascribed to the La–O stretching vibration and another of them is A1g peak at 201 cm−1 assigned to the La–O bending vibration.35 From Fig. 8(B), it is obvious that there is a negative shift of the Eg peak displayed for La2O3 in the hybrid (402 cm−1), compared with that of the pure La2O3. It implies that a change in covalence band of La2O3, which maybe result from the transferred electron from the carbon to La2O3, impacts the La–O stretching vibration peak.
 | ||
| Fig. 8 (A) Raman spectra for the pure carbon and the La2O3/C hybrid; (B) Raman spectra for the pure La2O3 and the La2O3/C hybrid. | ||
For the hexagonal La2O3 oxide, it has the A-type structure of rare-earth oxides and lanthanum cations locate in the center of the octahedral interstices formed by oxygen anions.25 According to this model, the eg orbital originates from a degenerate orbital splitted by the La 5d orbital of La2O3.25 The schematic crystal field splitting and occupations of the La3+ 5d and O 2p electrons in octahedral environments are given in Fig. 9(A). For La 5d orbital, the orbital energy of eg is much higher than the t2g, where eg and t2g are the two degenerate orbitals splitted by the La 5d orbital.36 For O 2p orbital, the strong hybridization of the La3+ 5d states with O 2p states leads to the existence of the filled O 2p orbitals with low energy, while a weak hybridization of the La3+ 5d states with O 2p states results in the unoccupied O 2p orbitals with high energy in this crystal field.25 Thus, due to the electron transfer from the carbon to the La2O3 shown in Fig. 9(B), the transferred electron (π electrons with high energy) from the carbon, may occupy the unfilled eg orbital or the unoccupied O 2p with high energy in La2O3. However, owing to the presence of the internal electron transition (O 2p → La 5d) in La2O3,36 the majority of the transferred electrons from carbon will occupy the empty eg orbital of La3+ atoms, as shown in Fig. 9(B). Such eg orbital filling in La2O3 phase has a good effect on the enhanced electro-catalytic performance of the hybrids toward ORR.
 | ||
| Fig. 9 (A) Schematic crystal field splitting and occupations of the La 5d electrons in octahedral environments; (B) schematic illustration of the electron transfer from the carbon to the eg orbital of the La2O3 in La2O3/C hybrid. | ||
As confirmed by Suntivich et al.37 the presence of a certain amount of eg electron (eg ≈ 1) for transition metal ions is remarkably sensitive to chemical adsorption of active oxygen, that is, the first step and rate-determining step of the ORR. More important, the filled eg electron can not only promote adsorption of active oxygen on the oxide surface but desorption of the surface-adsorbed hydroxide (OHad−) in solution, which contributes to trigger the ORR process.37 Therefore, such electron transferred from the carbon to the eg orbital of La2O3, through the covalent C–O–La bonds in this La2O3/C hybrid, may contribute its enhanced electro-catalytic activity toward the ORR. So, through modulating of metallic element d orbit via chemical interaction between the oxide and carbon in their hybrid, it may be a feasible and effective means to improve electrochemical performance of oxide-based catalysts.
4. Conclusions
In summary, we synthesized a set of La2O3/C hybrids with different La2O3 loadings via chemical precipitation of LaCl3 and CO(NH2)2 and followed by calcination at 750 °C. The obtained hexagonal La2O3 nanoparticles are all evenly dispersed on the surface of the carbon. The formation mechanism of La2O3 nanoparticles on carbon is proposed. And the electrochemical measurements in an alkaline solution show that catalytic activities for the hybrid samples display a volcano-type trend along with La2O3 loadings, where the peak locates at the La2O3/C catalyst with an oxide loading of 80 wt%, which is due to the maximum content of covalent C–O–La bonds at the interface between the carbon and oxide. Besides, the La2O3/C catalyst also exhibits a superior durability compared with a commercial Pt/C (E-TEK). For the La2O3/C hybrid, the observed electron, transferred from the carbon to the oxide through the covalent C–O–La bonds, would occupy the unfilled eg orbital of La3+ atoms in La2O3 phase. Such eg orbital filling greatly influence adsorption of active oxygen and desorption of the surface-adsorbed hydroxide (OHad−) on the oxide surface, which contributes to the enhanced ORR catalytic performance. This information may be useful to achieve hybrid catalysts composed by rare earth oxides and carbon as efficient ORR catalysts.Acknowledgements
This work was supported by National Natural Science Funds of China (Grant No. 51572013, 51432003).References
- A. Zhamu, G. Chen, C. Liu, D. Neff, Q. Fang, Z. Yu, W. Xiong, Y. Wang, X. Wang and B. Z. Jang, Energy Environ. Sci., 2012, 5, 5701–5707 CAS.
- G. Girishkumar, B. McCloskey, A. C. Luntz, S. Swanson and W. Wilcke, J. Phys. Chem. Lett., 2010, 1, 2193–2203 CrossRef CAS.
- F. Cheng, Y. Su, J. Liang, Z. Tao and J. Chen, Chem. Mater., 2010, 22, 898–905 CrossRef CAS.
- P. Tan, Z. H. Wei, W. Shyy and T. S. Zhao, Appl. Energy, 2013, 109, 275–282 CrossRef CAS.
- P. Tan, W. Shyy, Z. H. Wei, L. An and T. S. Zhao, Electrochim. Acta, 2014, 147, 1–8 CrossRef CAS.
- R. D. McKerracher, C. Alegre, V. Baglio, A. S. Aricò, C. Ponce de León, F. Mornaghini, M. Rodlert and F. C. Walsh, Electrochim. Acta, 2015, 174, 508–515 CrossRef CAS.
- C. Su, T. Yang, W. Zhou, W. Wang, X. M. Xu and Z. P. Shao, J. Mater. Chem. A, 2016, 4, 4516–4524 CAS.
- L. M. Dai, Y. H. Xue, L. Qu, H. J. Choi and J. B. Baek, Chem. Rev., 2015, 115, 4823–4892 CrossRef CAS PubMed.
- I. Karimzadeh, M. Aghazadeh, B. Safibonab, M. R. Ganjali and S. Dalvand, Russ. J. Electrochem., 2015, 51(3), 263–270 CrossRef CAS.
- T. Xia, J. P. Wang, N. Lin, L. H. Huo, H. Zhao and G. Mountrichas, J. Alloys Compd., 2010, 507, 245–252 CrossRef CAS.
- Q. Mu and Y. Wang, J. Alloys Compd., 2011, 509, 396–401 CrossRef CAS.
- Z. Barandiarán and L. Seijo, J. Chem. Phys., 2003, 119, 3785–3790 CrossRef.
- K. Liu, Y. Lei and G. Wang, J. Chem. Phys., 2013, 139, 204306 CrossRef PubMed.
- D. J. IIett and M. Salful Islam, J. Chem. Soc., Faraday Trans., 1993, 89, 3833–3839 RSC.
- X. X. Zhang, Q. Q. Xiao, Y. X. Zhang, X. Jiang, Z. Y. Yang, Y. F. Xue and Y. M. Yan, J. Phys. Chem. C, 2014, 118, 20229–20237 CAS.
- W. W. Gu, J. J. Liu, M. A. Hu, F. Wang and Y. Song, ACS Appl. Mater. Interfaces, 2015, 7, 26914–26922 CAS.
- H. C. Liu, J. J. Liu, W. W. Song, F. Wang and Y. Song, Mater. Lett., 2015, 139, 447–450 CrossRef CAS.
- T. F. Li, J. J. Liu, X. M. Jin, F. Wang and Y. Song, Electrochim. Acta, 2016, 198, 115–126 CrossRef CAS.
- J. Hu, L. N. Shi, Q. N. Liu, H. Huang and T. F. Jiao, RSC Adv., 2015, 5, 92096–92106 RSC.
- J. J. Liu, X. M. Jin, W. W. Song, F. Wang, N. Wang and Y. Song, J. Catal., 2014, 35, 1173–1188 CrossRef CAS.
- S. Kumar, A. K. Ojha, D. Patrice, B. S. Yadav and A. Materny, Phys. Chem. Chem. Phys., 2016, 18, 11157–11167 RSC.
- Y. G. Zhao, J. J. Liu, Y. H. Zhao, F. Wang and Y. Song, J. Mater. Chem. A, 2015, 3, 20086–20091 CAS.
- X. M. Ge, A. Sumboja, D. Wuu, T. An, B. Li, F. W. Thomas Goh, T. S. Andy Hor, Y. Zong and Z. L. Liu, ACS Catal., 2015, 5, 4643–4667 CrossRef CAS.
- M. Islam, J. Ilett and S. Parker, J. Phys. Chem., 1994, 98(38), 9637–9641 CrossRef CAS.
- X. L. Xu, Z. H. Chen, Y. Li, W. K. Chen and J. Q. Li, Surf. Sci., 2009, 603, 653–658 CrossRef CAS.
- P. Song, Q. Wang and Z. X. Yang, Sens. Actuators, B, 2009, 141, 109–115 CrossRef CAS.
- D. Kumar, M. A. Rub, M. Akram and K. ud-Din, J. Colloid Interface Sci., 2014, 418, 324–329 CrossRef CAS PubMed.
- M. L. Bhaisare, S. Pandey, M. S. Khan, A. Talib and H. F. Wu, Talanta, 2015, 132, 572–578 CrossRef PubMed.
- Y. D. Wang, C. L. Ma, X. D. Sun and H. D. Li, Inorg. Chem. Commun., 2002, 5, 751–755 CrossRef CAS.
- J. Sunarso, J. Torriero and W. Zhou, J. Phys. Chem. C, 2012, 116, 5827–5834 CAS.
- J. Z. Liu, J. J. Liu, W. W. Song, F. Wang and Y. Song, J. Mater. Chem. A, 2014, 2, 17477–17488 CAS.
- C. V. Ramana, R. S. Vemuri and V. V. Kaichev, ACS Appl. Mater. Interfaces, 2011, 3, 4370–4373 CAS.
- H. C. Liu, J. J. Liu, F. Wang and Y. Song, RSC Adv., 2015, 5, 90785–90796 RSC.
- K. S. Subrahmanyam, A. K. Manna, S. K. Pati and C. N. R. Rao, Chem. Phys. Lett., 2010, 497, 70–75 CrossRef CAS.
- J. Zarembowitch, et al., Phys. Status Solidi B, 1979, 94, 249–256 CrossRef CAS.
- A. B. Altman, J. I. Pacold, J. Wang, W. W. Lukens and S. G. Minasian, Dalton Trans., 2016, 45, 9948–9961 RSC.
- J. Suntivich, H. A. Gasteiger, N. Yabuuchi, H. Nakanishi, J. B. Goodenough and Y. Shao-Horn, Nat. Chem., 2011, 3, 546–550 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2016 |