
Tuning Ni nanoparticles and the acid sites of silica-alumina for liquefaction and hydrodeoxygenation of lignin to cyclic alkanes†
Abstract
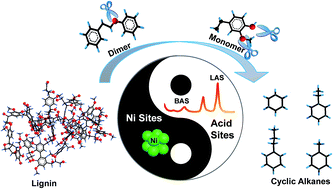
A facile and effective method for the one-pot hydrodeoxygenation of enzymatic lignin to C6–C9 cycloalkanes is reported in liquid dodecane with 100 C% selectivity (approaching 50 wt% yield). The method enables 80 wt% lignin conversion by using Ni catalyst-supported amorphous silica-alumina (ASA) at 300 °C in the presence of 6 MPa H2. The crucial factors to achieve direct lignin hydrodeoxygenation are the suitable balance in solvent selection and the design of active sites in the solvent liquid phase. The activity of Ni nanoparticles in dodecane leads to higher efficiency in the deconstruction of external C–O bonds in lignin. The consumption of lignin shifts the equilibrium of lignin solubility and weakens the impact of the relatively poor lignin solubility in dodecane for lignin depolymerization. The key element in controlling the activity of Ni-based catalysts is the specific external surface areas of diverse supports as well as the sizes of metallic Ni sites. This is probably because of the high external surface areas that can provide good contact opportunities for Ni sites in the lignin macromolecule. The efficient contact of active sites in the polymer reactant is the most important factor for such solid–solid reactions. The size and distribution of active Ni sites as well as the specific surface areas of Ni/ASA as modified by the different deposition precipitation times, reduction temperatures, and Ce additives can greatly affect the ability of a metal to attack the external C–O bonds of lignin. Furthermore, the acidity of the support (especially Brönsted acid sites) as modified by the Si/Al ratio of ASA significantly enhances the capabilities and alters the electronic structures of Ni nanoparticles for cleavage of the C–O linkages of lignin. This suggests that the synergy of acid and metal sites can be subtly tailored to strengthen the catalytic performance of Ni metallic sites. In addition, the presence of acidic sites catalyzes the dehydration of cyclic alcohols intermediates and facilitates the hydrodeoxygenation of the derived phenolic fragments to cyclic alkanes.
 Please wait while we load your content...
Please wait while we load your content...

