
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Facile high-yield synthesis of unsymmetric end-off compartmental double Schiff-base ligands: easy access to mononuclear precursor and unsymmetric dinuclear complexes†
Markus
Schmidt
,
Helmar
Görls
and
Winfried
Plass
*
Institut für Anorganische und Analytische Chemie, Friedrich-Schiller-Universität Jena, Humboldtstrasse 8, 07743 Jena, Germany. E-mail: sekr.plass@uni-jena.de; Fax: +49 3641 948132; Tel: +49 3641 948130
First published on 3rd August 2016
Abstract
A straightforward and easy to handle two-step synthetic route for unsymmetric double Schiff-base ligands is presented. The isolated intermediate single Schiff-base precursor ligands H2sc-difo and H2tsc-difo were derived by condensation of 2,6-diformyl-4-methylphenol with semicarbazide and thiosemicarbazide, respectively. Further reaction of the precursor ligands with different amine components, including both aliphatic and aromatic examples, allows the synthesis of ditopic unsymmetric double Schiff-base ligands in high yields and purity. As aliphatic cases we used (2-aminoethyl)bis(2-pyridylmethyl)-amine (H2sc-hydra and H2tsc-hydra) and 2-(aminomethyl)-pyridine (H2sc-ampy and H2tsc-ampy), whereas 2-aminophenol was used as an aromatic sample (H3sc-amph and H3tsc-amph). The overall synthetic route allows for the preparation of the employed ligands in large scale. To explore the coordination capabilities of the reported ligand systems a mononuclear nickel(II) complex [Ni(tsc-difo)PPh3] and a homodinuclear zinc(II) complex [Zn2(tsc-hydra)(OAc)2] were synthesized with the single Schiff-base precursor ligand H2tsc-difo and the double Schiff-base ligands H2tsc-hydra, respectively. Both complexes crystallized in the monoclinic space group P21/n. For [Ni(tsc-difo)PPh3] a square-planar geometry is found for nickel(II) ion with H2tsc-difo acting as a tridentate ligand. Whereas the structure of complex [Zn2(tsc-hydra)(OAc)2] reveals two zinc(II) ions in distinctly different coordination geometry, one in distorted octahedral coordination located in the bispyridine based binding pocket with [N4O2] donor set and the other zinc(II) ion in a distorted square-pyramidal coordination given by the thiosemicarbazone based binding pocket with [NO3S] donor set.
Introduction
Bimetallic active sites are widely spread in metalloenzymes of numerous biological systems for which examples have been isolated and structurally characterized.1,2 As a prominent feature for such active sites the metal centers are usually embedded in distinctly different binding pockets.3 The resulting lack of symmetry can be due to different donor atoms involved as well as the coordination number and geometry at the metal sites.4 This enables the two metal centers to perform different tasks within the enzymatic activity. Examples of such metalloenzymes range from homodinuclear cases like urease5,6 (Ni2), metallo-β-lactamase7 (Zn2), and the dioxygen carrier hemerythrin8 (Fe2) towards heterobimetallic metallobiosites as in purple acid phosphatases9 (Fe/Zn), copper–zinc superoxide dismutase10 (Cu/Zn), and arylamine oxygenase AurF11,12 (Fe/Mn). Moreover, bimetallic complexes also play an important role in catalytic processes and novel abiotic molecule transformations. The concept of cooperative reactivity based on bimetallic catalysts13–15 has lead to interesting applications such as asymmetric rearrangements,16 ring opening reactions,17 hydroalkoxylation of alkynes,18 palladium-assisted C–X bond formation19 and polymerizations.20,21Over the past decades compartmental ligands capable of coordinating multiple metal ions have been an intense area of research.22,23 Among these supporting scaffolds unsymmetric ligands deliberately providing unequal binding pockets for metal ions are of particular interest.24–29 Several approaches have been proposed to synthesize such unsymmetric ligands that utilize multi-step synthetic routes typically starting from predesigned unsymmetric bridging units.30–44 Although Schiff-base ligands have been found to offer excellent potential as their coordination ability and applications of related complexes are concerned, their utilization in the design of unsymmetric ditopic ligand systems is still limited.45,46
In this context we have reported a versatile synthetic approach towards double Schiff-base ligands which is based on stepwise Schiff-base condensation utilizing the symmetric dicarbonyl bridge 2,6-diformyl-4-methylphenol (dfc) and two different primary amines (see Fig. 1).47 The related end-off compartmental ligands systems can be utilized for directed synthesis of heterobimetallic complexes.48 However, the crucial step in the synthesis of these double Schiff-base ligands is the separation of the monocarbonyl precursor ligand from the symmetric side products by size-exclusion chromatography which has some drawbacks. Primarily this considerably limits the scale of reaction to the lower millimolar range and due to the prolonged times needed during reaction and separation this can lead to a small portion of undesired re-symmetrization due to imine metathesis.49
 | ||
| Fig. 1 General scheme for the synthesis of unsymmetric double Schiff bases indicating the possible formation of symmetric side products. | ||
Here we describe a straightforward and easy to handle large scale synthetic approach towards unsymmetric double Schiff bases via isolated intermediate single Schiff-base precursors based on thiosemicarbazide and semicarbazide as amine components. Schiff bases containing the latter are not only known for their good coordination abilities50 but also for the formation of complexes with antibacterial, antiviral, anticancer, and other biological properties.51–53
Experimental
Instrumentation
The NMR spectra were measured with Bruker Avance 400 MHz and 600 MHz spectrometers. For recording the IR spectra a Bruker IFS55/Equinox spectrometer equipped with a diamond ATR unit was used. The UV/Vis data were collected on a Cary Varian 5000 UV/Vis/NIR spectrometer. Mass spectra were carried out with the help of a MAT95XL Finnigan instrument. The elemental analyses were determined with a VARIO EL III and LECO CHN/932 elemental analyzers.Materials
The starting materials 2,6-diformylcresol (dfc)54 and (2-aminoethyl)bis(2-pyridylmethyl)amine55 were prepared according to the methods described in literature. All other chemicals were purchased from commercial suppliers and applied as received, except for the solvents which were purified and distilled prior to use.Synthesis of proligands
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N, H-2), 10.03 (s, 1H, HCO, H-10), 10.91 (s, 1H, OH), 11.51 (s, 1H, NH). 13C{1H} NMR (100 MHz, DMSO-d6, δ in ppm, see Fig. S2†): 19.7 (C9), 121.8 (C7), 122.1 (C3), 129.1 (C5), 134.2 (C4), 134.3 (C6), 137.1 (C2), 156.7 (C8), 177.9 (C1), 195.7 (C10). MS (DEI, m/z, rel. int. in %): 237 (70, M+), 162 (100), 133 (32), 106 (30), 76 (52). FT-IR: see Fig. S3.†
N, H-2), 10.09 (s, 1H, HCO, H-10), 10.38 (s, 1H, NH), 11.13 (s, 1H, OH). 13C{1H} NMR (100 MHz, DMSO-d6, δ in ppm, see Fig. S4†): 19.7 (C9), 121.9 (C7), 122.2 (C3), 128.8 (C5), 132.3 (C6), 134.1 (C4), 135.5 (C2), 156.3 (C1), 156.4 (C8), 194.6 (C10). MS (DEI, m/z, rel. int. in %): 237 (98, M+), 193 (67), 162 (100), 133 (82), 106 (57). FT-IR: see Fig. S5.†
N, H-2), 10.03 (s, 1H, HCO, H-10), 10.91 (s, 1H, OH), 11.51 (s, 1H, NH). 13C{1H} NMR (100 MHz, DMSO-d6, δ in ppm, see Fig. S2†): 19.7 (C9), 121.8 (C7), 122.1 (C3), 129.1 (C5), 134.2 (C4), 134.3 (C6), 137.1 (C2), 156.7 (C8), 177.9 (C1), 195.7 (C10). MS (DEI, m/z, rel. int. in %): 237 (70, M+), 162 (100), 133 (32), 106 (30), 76 (52). FT-IR: see Fig. S3.†
N, H-2), 10.09 (s, 1H, HCO, H-10), 10.38 (s, 1H, NH), 11.13 (s, 1H, OH). 13C{1H} NMR (100 MHz, DMSO-d6, δ in ppm, see Fig. S4†): 19.7 (C9), 121.9 (C7), 122.2 (C3), 128.8 (C5), 132.3 (C6), 134.1 (C4), 135.5 (C2), 156.3 (C1), 156.4 (C8), 194.6 (C10). MS (DEI, m/z, rel. int. in %): 237 (98, M+), 193 (67), 162 (100), 133 (82), 106 (57). FT-IR: see Fig. S5.†
Synthesis of double Schiff bases
A solution of (2-aminoethyl)bis(2-pyridylmethyl)amine in methanol (400 mL) was added dropwise over a period of 24 h to a yellow suspension of one equivalent of the corresponding proligand in methanol (100 mL). The reaction mixture immediately turned to an orange to red color and became a clear solution at the end of the addition. Subsequently the solvent was removed under reduced pressure at a temperature of 40 °C by means of a rotary evaporator. The residual orange-red solid was washed several times with diethyl ether to afford the unsymmetric double Schiff-base ligand as orange to red powder.Synthesis of complexes
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 155), 427 (8770, shoulder), 386 (10349), 370 (8458, shoulder), 340 (5907), 303 (18189). MS (Micro-ESI pos., m/z, rel. int. in %): 577.8 (66, [M + Na]+), 413 (26), 333 (58), 301 (100), 263 (62, P(Ph)3). FT-IR: see Fig. S20.†
155), 427 (8770, shoulder), 386 (10349), 370 (8458, shoulder), 340 (5907), 303 (18189). MS (Micro-ESI pos., m/z, rel. int. in %): 577.8 (66, [M + Na]+), 413 (26), 333 (58), 301 (100), 263 (62, P(Ph)3). FT-IR: see Fig. S20.†
X-ray crystallographic studies
The intensity data were collected on a Nonius KappaCCD diffractometer, using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). Data were corrected for Lorentz and polarization effects. Absorption was taken into account on a semi-empirical basis using multiple-scans.56–58 The structures were solved by direct methods (SHELXS)59 and refined by full-matrix least squares techniques against Fo2 (SHELXL-97).59 In the crystal structure of [Zn2(tsc-hydra)(OAc)2]·MeCN·H2O one of the pyridyl groups was found to be slightly disordered on two positions (ratio of site occupation factors is about 6 to 1). The hydrogen atoms of the amine group (N1) and the water molecule (O1W) in the crystal structure of the zinc complex were located by difference Fourier synthesis and refined isotropically. All other hydrogen atom positions were included at calculated positions with fixed thermal parameters. All non-hydrogen and non-disordered atoms were refined using anisotropic thermal parameters. Crystallographic data as well as structure solution and refinement details for [Ni(tsc-difo)PPh3]·2MeOH and [Zn2(tsc-hydra)(OAc)2]·MeCN·H2O are summarized in Table S1.†Results and discussion
Mono Schiff-base proligands
First attempts to synthesize the desired mono Schiff-base proligand H2tsc-difo depicted in Fig. 2 utilized the slow addition of a diluted thiosemicarbazide (tsc) solution to a concentrated solution of dfc in methanol over a period of one week. Although this successfully led to the formation of the desired mono Schiff base, the product contained a considerable amount of the symmetric double Schiff base as byproduct. Since the resulting product mixture was only sparingly soluble in acetonitrile and chloroform, standard recrystallization could not be applied. However, attempts by Soxhlet extraction with chloroform over a period of four days could successfully be utilized to separate the unsymmetric proligand from the undesired symmetric side product. H2tsc-difo was extracted from the crude reaction product and precipitated in pure form as a yellow solid which is evident by comparison of the NMR spectra before and after extraction (Fig. S24†). Based on these observations we supposed that under conditions of continuous dilution over the whole period of reaction, with respect to the amine component tsc, it should be possible to generate solely H2tsc-difo without any symmetric byproduct. | ||
| Fig. 2 Mono Schiff-base proligands. | ||
Along this line it turned out to be beneficial that dfc shows a solubility in acetonitrile which is by far larger than that of tsc. This allows during the reaction to simultaneously maintain a rather high dfc concentration paired with a situation of continuous dilution with respect to tsc. The desired idealized reaction conditions should combine a steady but very small intake of tsc to a highly concentrated dfc reaction solution. These conditions, best described as slow solution transport, can be achieved by a simple reaction setup which utilizes the solvent extraction of tsc from a fritted glass filter by acetonitrile under reflux conditions as depicted in Fig. S1.† To maintain the desired dilution conditions with respect to the amine component throughout the whole reaction time a 1.5 stoichiometric excess of the carbonyl component dfc was used. Within a period of a about two days the complete amount of tsc was eluted into the reaction mixture and the proligand H2tsc-difo was formed as yellow solid precipitate insoluble in the reaction mixture. As the solubility of the mono Schiff-base proligand increases with temperature, which would favor the formation of the undesired symmetric byproduct, the reaction temperature was kept below approximately 40 °C by reducing the pressure in the reaction vessel and thereby lowering the boiling point of the reaction solvent acetonitrile. The remaining excess of dfc can easily be recovered from the final solid reaction product by washing the yellow powder several times with either chloroform or dichloromethane. The scale-up of this procedure was optimized to produce the proligand in gram scale and over 90% yield. The basic limiting factor is the solubility of the dfc starting material in the given reaction volume.
This synthetic strategy should be transferable to the reaction of dfc with semicarbazide (sc). However, the varied solubility properties of semicarbazide and the fact that it is commercially available as hydrochloride, require some modifications of the applied procedure. In this case it was necessary to utilize dichloromethane as reaction solvent, since the solubility of semicarbazide hydrochloride is too high in acetonitrile to prevent the formation of the undesired symmetric side product. Moreover, to generate the free semicarbazide triethylamine was added as base to the reaction mixture. As in the previous case the semicarbazide is slowly eluted into the reaction flask. The product H2sc-difo (see Fig. 2) is formed as a pale yellow precipitate which can be isolated from the crude reaction mixture by filtration and subsequent washing with dichloromethane. However, the yield is slightly lower compared to the previous case of the thiosemicarbazone derivative as H2sc-difo possesses a somewhat better solubility and is therefore partially lost during the washing process.
Both proligands H2sc-difo and H2tsc-difo can be obtained in high yields as pure solid materials. This synthetic route provides a large scale access to these versatile ligand systems which can be utilized for further derivatization in various ways. The available free carbonyl group of the proligands can be functionalized with other amine components to yield the desired unsymmetric double Schiff bases. Alternatively the proligands can directly used for coordination of metal ions leading to mononuclear complexes which in turn could be further functionalized at the remaining carbonyl group.
Double Schiff bases
Starting from the proligands H2sc-difo and H2tsc-difo unsymmetric double Schiff bases can be derived by condensation in methanol with the appropriate amine components. To probe the synthetic versatility of this approach we utilized three different primary amines including aliphatic and aromatic examples which vary the number and nature of the introduced donor atoms, namely (2-aminoethyl)bis(2-pyridylmethyl)amine, 2-(aminomethyl)-pyridine and 2-aminophenol. These reactions were performed under heterogeneous conditions since the proligands were only sparingly soluble in methanol. The resulting double Schiff bases depicted in Fig. 3 are obtained in high yield and purity without any indication of imine metathesis during the course of reactions.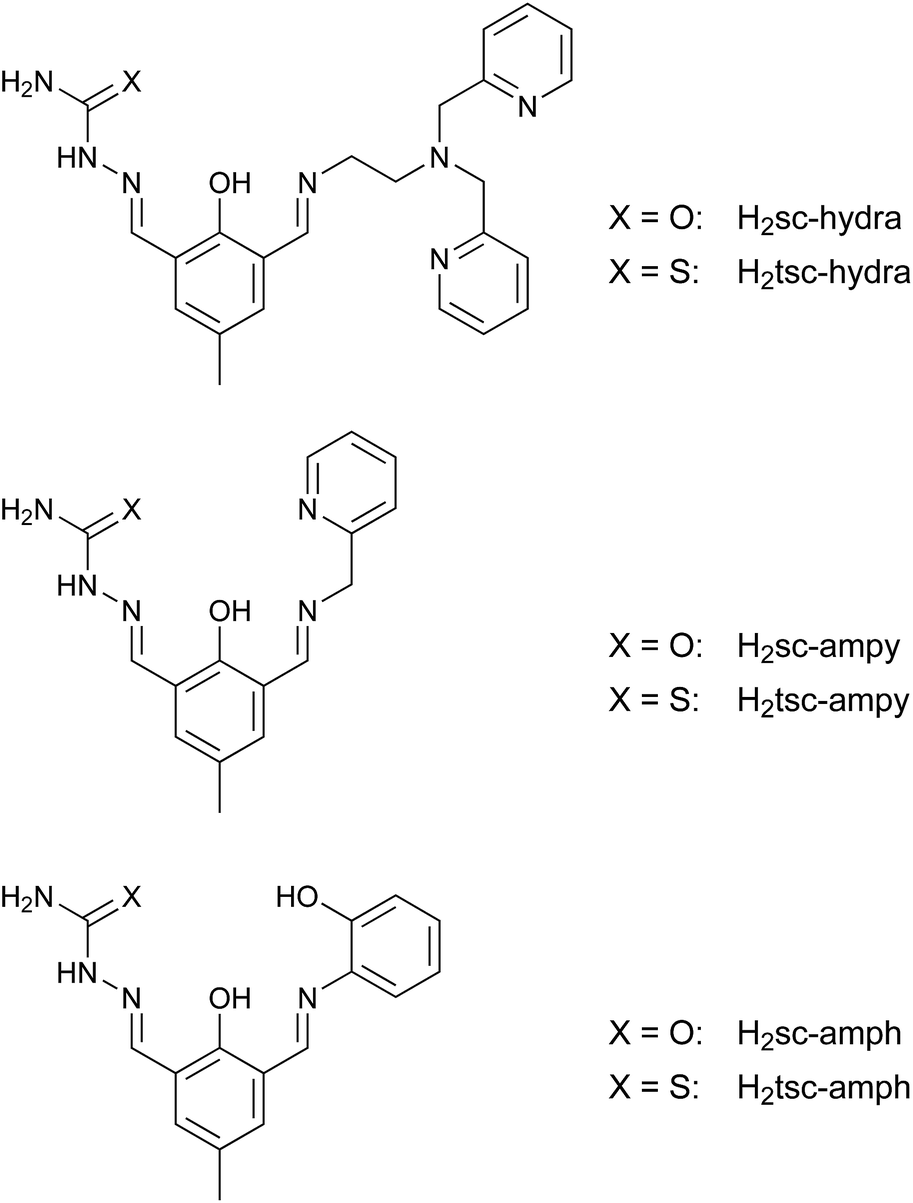 | ||
| Fig. 3 Synthesized double Schiff-base ligands based on the proligands H2sc-difo and H2tsc-difo. | ||
All these potential ligands contain two distinct binding pockets for the coordination of metal ions. According the nature of the second amine group introduced these ligands fall into two groups with either an additional flexible or rigid donor fragment, besides the generally present semicarbazone or thiosemicarbazone moiety which provides a binding pocket with rigid tridentate donor set.
Selected 1H NMR data for the double Schiff bases are summarized in Table 1. The resonances of the methine protons assigned to the imine groups are observed in the usual range and show the expected shielding effect when compared to the corresponding resonance of the proligands (H-10). These data further indicate the presence of strong hydrogen-bonding interaction between the hydroxy group of the bridging aromatic moiety and the adjacent imino group as depicted in Fig. 4. This leads to a resonance-assisted hydrogen bond N–H⋯O which is well known for Schiff bases containing salicylidene moieties.60,61 As a consequence the corresponding proton resonance is considerably shifted downfield to values of more than 14 ppm in comparison to the resonances observed for the proligands H2sc-difo and H2tsc-difo at around 11 ppm lacking this interaction. A comparison of selected 13C NMR data for the proligands and double Schiff bases are summarized in Table S2.†
| Ligand | N–H⋯O | OH | H-10 | H-2 | NH | NH2 |
|---|---|---|---|---|---|---|
| H2sc-difo | — | 11.13 | 10.09 | 8.13 | 10.38 | 6.52 |
| H2tsc-difo | — | 10.91 | 10.03 | 8.35 | 11.51 | 8.08 |
| 8.24 | ||||||
| H2sc-hydra | 14.17 | — | 8.45 | 8.20 | 10.28 | 6.50 |
| H2tsc-hydra | 14.37 | — | 8.47 | 8.47 | 11.48 | 8.02 |
| 8.18 | ||||||
| H2sc-ampy | 14.02 | — | 8.69 | 8.17 | 10.62 | 6.48 |
| H2tsc-ampy | 14.18 | — | 8.69 | 8.39 | 11.46 | 7.99 |
| 8.17 | ||||||
| H3sc-amph | 14.48 | 9.84 | 8.93 | 8.25 | 10.29 | 6.50 |
| H3tsc-amph | 14.62 | 9.85 | 8.95 | 8.46 | 11.48 | 8.00 |
| 8.17 |
 | ||
| Fig. 4 Tautomerization between the hydroxy-imine and keto-amine forms of the free double Schiff-base ligands in solution. | ||
The observed NMR data is consistent with a preserved solution behavior for the semicarbazone and thiosemicarbazone moieties within the two series of mono and double Schiff bases. A usually observed structural feature for these class of compounds is the almost planar CN–NH–CX–NH2 backbone with a trans arrangement of the azomethine and the X (O, S) atom as depicted in Fig. 5.62 For the thiosemicarbazones this is known to lead to restricted rotation of the NH2 group,63,64 which might be attributed to an increased contribution of the zwitterionic resonance form (see Fig. 5) as well as the possible hydrogen bonding of one of the NH2 protons with the azomethine nitrogen atom. This is obvious from two independent resonances for the NH2 protons within the thiosemicarbazone series which are both shifted downfield with respect to the corresponding semicarbazones.
 | ||
| Fig. 5 Mesomeric forms for the thiosemicarbazone based ligands H2tsc-difo, H2tsc-hydra, H2tsc-ampy and H3tsc-amph preventing free rotation of the NH2 group. | ||
For the double Schiff base H2tsc-hydra this effect has been more closely examined by variable temperature 1H NMR spectroscopy with the resulting spectra depicted in Fig. 6. The two resonances observed for the NH2 group at room temperature coalesce upon increasing the temperature. From the spectral data the free energy of activation ΔG‡ can be estimated65 to 69.9 kJ mol−1 which is in good agreement with reported values.66
 | ||
| Fig. 6 Variable temperature 1H NMR spectra for H2tsc-hydra measured at a spectrometer frequency of 400 MHz showing the coalescence for the proton resonances of the NH2 group (marked peaks). | ||
Ni(II) complex with the proligand H2tsc-difo
The mononuclear complex [Ni(tsc-difo)PPh3] was synthesized by slow addition of a methanol solution of nickel(II) perchlorate hexahydrate to a slurry of the mono Schiff-base proligand H2tsc-difo containing one equivalent of triphenylphosphane as potential coligand and two equivalents of triethylamine as base. Upon complexation of the nickel ions a color change from yellow to dark red is observed and the reaction mixture turns in a clear red solution. The observed color indicates a square-planar coordination geometry at the nickel(II) ion, as expected for a coordination of the strong phosphane ligand. This is confirmed by the UV/Vis spectrum of the isolated crystalline complex which is typical for a square-planar nickel(II) complex67 with thiosemicarbazone ligand featuring additional charge-transfer bands.64,68–70The well resolved 1H NMR spectrum of [Ni(tsc-difo)PPh3] measured for a dichloromethane-d2 solution is consistent with the expected diamagnetic ground state for a square-planar configuration at the nickel(II) center. The protons related to the resonance observed at 4.79 ppm are affected by H–D exchange and consequently assigned as the NH2 group of the thiosemicarbazone moiety, which is well within the usually observed range for anionic thiosemicarbazones coordinated to metal ions.50 The fact that in contrast to the free ligand only one resonance is observed for the NH2 group can be attributed to missing intramolecular hydrogen bonding due to an altered configuration of the thiosemicarbazone moiety in the complex. The protons related to the second resonance showing H–D exchange characteristic observed at 3.42 ppm are assigned to methanol molecules which are included in the crystal structure. Based on the integration of the resonance peak this corresponds to a methanol content of about 0.7 equivalents per complex unit which is in good agreement with the elemental analysis of the crystalline material dried in vacuo. The resonance of the triphenylphosphane protons are observed in the usual range,50 with the multiplet at 7.43 ppm being superimposed with the resonance of the ring proton H-6 of the 2,6-diformylcresol moiety. The resonance of the azomethine proton (H-2) is slightly shifted upfield upon coordination to 8.21 ppm and is split to a doublet due to coupling with the phosphorus atom of the triphenylphosphane in trans position at the nickel ion with a rather large coupling constant 4JPH = 8.9 Hz.71 The observation of a rather large 4JPH coupling indicates the presence of a highly rigid structural arrangement of the involved bonding backbone in solution.72 The 31P NMR resonance for [Ni(tsc-difo)PPh3] observed at 20.63 ppm is consistent with the usual range for coordination shifts observed for triphenylphosphane coordinated to nickel(II) ions.73
Crystals of [Ni(tsc-difo)PPh3] were obtained by slow evaporation of the solvent from the filtrated reaction mixture and were found to crystallize in the monoclinic space group P21/n together with two molecules of methanol. The molecular structure of the neutral complex is depicted in Fig. 7 (selected bond angle see Table S3†) and confirms the square-planar geometry at the nickel(II) center. The almost ideal square-planar coordination environment is given by the donor atoms O1, N3 and S of the proligand and P of the triphenylphosphane with a deviation of the nickel atom from the mean plane by only 2 pm. The bond lengths at the nickel(II) ion are within the expected range for similar complexes and consistent with the deprotonated thiolate form of the thiosemicarbazone fragment expected for a neutral complex.64,70,71 The bite angles for the five- and six-membered chelate rings of the tridentate thiosemicarbazone ligand are 95.1° (O1–Ni–N3) and 87.3° (N3–Ni–S1), respectively. The two planar chelate rings show a dihedral angle of 12°, which is due to a slight rotation about the C2–C3 bond.
 | ||
| Fig. 7 Molecular structure of [Ni(tsc-difo)PPh3]. Thermal ellipsoids are drawn with 50% probability level and hydrogen atoms attached to carbon are omitted for clarity. Pertinent distances (in pm): Ni–S 212.82(8), Ni–P 219.68(8), Ni–O1 185.8(2), Ni–N3 188.7(2), S–C1 175.2(3), N1–C1 135.6(4), N2–C1 130.5(4), N2–N3 139.9(3), N3–C2 130.3(4). | ||
The two additional molecules of methanol in the crystal structure are involved in hydrogen bonding between the NH2 group (N1) and the aldehyde moiety (O2) of a neighboring complex molecule leading to the formation of a chain along the crystallographic [![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 01] direction as depicted in Fig. 8. In addition NH/π interactions are observed between the second proton of the NH2 groups of the thiosemicarbazone moiety and the aromatic ring of an adjacent complex.74,75 This leads to a zig–zag chain like association along the [010] direction (see Fig. S25†).
01] direction as depicted in Fig. 8. In addition NH/π interactions are observed between the second proton of the NH2 groups of the thiosemicarbazone moiety and the aromatic ring of an adjacent complex.74,75 This leads to a zig–zag chain like association along the [010] direction (see Fig. S25†).
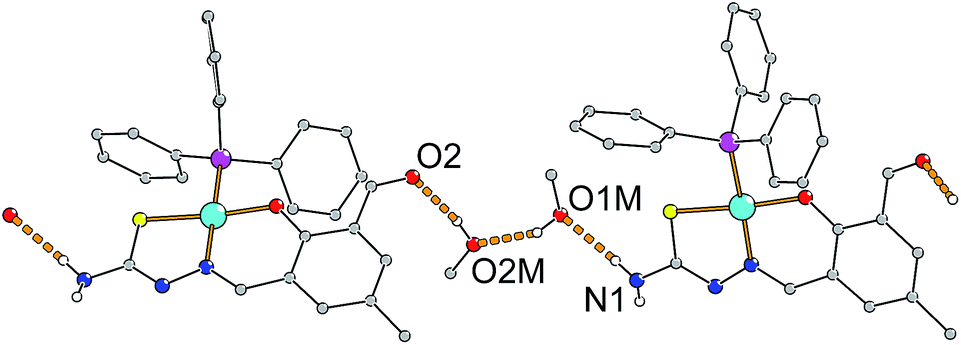 | ||
| Fig. 8 Hydrogen bonded chains in the crystal structure of [Ni(tsc-difo)PPh3]·2MeOH along the [01] direction. Pertinent distances (in pm): N1⋯O1M 291.6(5), O1M⋯O2M 269.8(5), O2⋯O2M 271.1(4). | ||
Dinuclear Zn(II) complex with H2tsc-hydra
The complex [Zn2(tsc-hydra)(OAc)2] was obtained from the reaction of two equivalents of zinc(II) acetate dihydrate and the double Schiff-base ligand H2tsc-hydra in a mixture of acetonitrile and methanol in the presence of triethylamine as base. The constitution of the complex was confirmed by analytical and spectroscopic methods. In particular the solution structure was studied by NMR spectroscopy.The 1H NMR spectra indicate significant changes upon coordination of the H2tsc-hydra to the zinc(II) ions with the observed resonances being generally broader than in the case of the free ligand. As in the case of the [Ni(tsc-difo)PPh3] the thiosemicarbazone moiety shows the typical features expected for the coordinated thiolate form with a singlet at 6.22 ppm for the protons of the NH2 group. The latter assignment was confirmed by H–D exchange experiments. The two pyridyl rings give rise to only one set of resonances consistent with the equivalence of the two coordinated pyridyl moieties. Similarly for the two acetate anions present in the complex only a single broad resonance at 1.65 ppm is observed indicating their chemical equivalence on the NMR time scale. The resonances for the methylene protons within the ethylenediamine bridge observed at 2.82 (C12) and 2.98 ppm (C11) show the basic trends upon coordination usually observed in the literature.48,76,77 However, in contrast to the free ligand and to an earlier reported dinuclear zinc complex with a similar ligand framework spin–spin coupling is not resolved.48 Moreover, the resonance for the methylene group (2.82 ppm) attached to the tertiary nitrogen donor appears as a very broad singlet with a width at half-height of about 125 Hz. On the other hand, for the methylene hydrogens of the pyridyl arms (C13 and C19) a well resolved AB pattern with a coupling constant of 16.8 Hz is observed.65 This is typical for a geminal coupling related to this type of coordination pocket and clearly indicates the stereochemical disparity of the involved hydrogen atoms.48,78
Crystals of [Zn2(tsc-hydra)(OAc)2] suitable for single crystal analysis were obtained by slow evaporation of the solvent. The neutral complex was found to co-crystallize with one additional molecule of water and acetonitrile as [Zn2(tsc-hydra)(OAc)2]·MeCN·H2O in the monoclinic space group P21/n. The molecular structure of the neutral complex is depicted in Fig. 9 (selected bond angle see Table S4†).
 | ||
| Fig. 9 Molecular structure of [Zn2(tsc-hydra)(OAc)2]. Thermal ellipsoids are drawn with 50% probability level and hydrogen atoms attached to carbon are omitted for clarity. Pertinent distances (in pm): Zn1–S 235.11(12), Zn1–O1 212.3(3), Zn1–O2 197.6(3), Zn1–O4 206.8(3), Zn1–N3 209.9(3), Zn2–O1 212.4(3), Zn2–O4 210.5(3), Zn2–N4 209.6(4), Zn2–N5 227.7(3), Zn2–N6 214.0(4), Zn2–N7 210.6(4). | ||
Two different binding modes are observed for the two coordinated zinc(II) ions. Zn1 is found in a five-coordinate square-pyramidal environment with an [NO3S] donor set whereas for Zn2 a six-coordinate octahedral geometry with [N4O2] donor set is observed. This is not surprising, as zinc(II) ions usually show no specific structural preferences. Therefore the overall structure can be expected as being governed by the donor properties and the flexibility of the two distinctly different binding pockets ligand system. The overall coordination of the zinc(II) ions is complemented by two additional acetate anions one in a μ,η1-bridging mode and the second one as a terminal monodentate ligand at zinc center located in the thiosemicarbazone pocket (Zn1).
The central Zn2O2 moiety is virtually planar within a deviation of less than 2 pm from the mean plane. The two corresponding basal coordination planes at the square-planar Zn1 center (S, O1, O4, and N3) and the octahedral Zn2 center (O1, O4, N4, and N5) show dihedral angles of 18° and 7° with the central Zn2O2 moiety, respectively. Interestingly the deviations of the zinc ions from the respective mean planes exhibit distinct differences (Zn1: 58 pm; Zn2: 3 pm) indicating considerable distortion within the basal coordination plane at the Zn2 center. This also reflects in the deviation of the nitrogen donor atoms N4 (−59 pm) and N5 (21 pm) from the mean plane of the central Zn2O2 moiety. The overall situation is depicted in Fig. 10.
 | ||
| Fig. 10 Representation of the coordinations spheres of the zinc(II) ions in [Zn2(tsc-hydra)(OAc)2]. The two depicted planes contain the common O1–O4 axis as hinge and correspond to the mean planes given by the (O1, O4, N5) and (S, O1, O4, N3) which create an angle of about 20°. Pertinent distances from planes (in pm): (O1, O4, N5) Zn2 12.8, N4 75.0; (S, O1, O4, N3) Zn2 45.9, N4 22.8. | ||
To accommodate the rigidity of the π conjugated part of the ligand backbone to the described distortion the bridging phenolate fragment is tilted with respect to the central Zn2O2 moiety. This corresponds to a rotation about the C2–C3 bond leading to a dihedral angle of around 16° (see Fig. S26†). Moreover, there is an additional source of distortion for the pyridyl based binding compartment of the ligand system around Zn2 which is due to the five-membered chelate rings leading to significant decrease of the trans angles with respect to an ideal octahedral geometry (O1–Zn2–N5 161.9°, O4–Zn2–N4 155.6°, N6–Zn2–N7 149.3°). As a consequence there is enough space opposite to the ligand backbone at the two zinc(II) ions to allow a coplanar orientation of the bridging acetate anion with respect to the central Zn2O2 moiety.
The supramolecular arrangement within the crystal structure is governed by hydrogen bonding between pairs of neutral complexes via the thiosemicarbazone fragment of adjacent molecules. Together with hydrogen bonding towards the additional co-crystallized water molecule this leads to the formation of chain-like aggregation of the complex molecules along the [100] direction as depicted in Fig. 11. Two such symmetry related chains buildup the crystal structure with relative orientations of their planar hydrogen paired ligand backbones aligned along the crystallographic (011) and (01) planes. The acetonitrile molecules are located between these chains such that they have hydrogen bonding contacts with neighboring chains of the same basic orientation (see Fig. S27†). The closest contact between chains of different orientation are π–π interactions of a pyridyl ring (N7) with the central aromatic ring of an adjacent complex molecule (see Fig. S28†). Together with additional packing effects due to the acetonitrile molecule the latter interaction leads to an out-of-plane distortion of the phenolate oxygen O1 (16 pm) and the methyl group C9 (18 pm) substituents at the central aromatic ring (C3 to C8).
 | ||
| Fig. 11 Representation of the hydrogen bonding interactions in the crystal structure of [Zn2(tsc-hydra)(OAc)2]·MeCN·H2O leading to a chain-like arrangement along the [100] direction. Pertinent distances (in pm): N1⋯N2 304.3(5), N2⋯O1W 295.7(6), O5⋯O1W 275.0(5), O1W⋯N1AN 298.4(9). | ||
Conclusions
A simple and straightforward synthetic approach towards unsymmetric double Schiff-base ligands on the basis of isolated intermediate mono Schiff-base precursors is presented. The first step of this synthetic route can generally be handled on a large scale when thiosemicarbazide and semicarbazide are used as amine components to generate the mono Schiff bases. These precursors can be utilized to synthesize a variety of unsymmetric end-off compartmental double Schiff-base ligands with both flexible and rigid donor sets. For two examples of the presented ligands their coordination behavior has been studied by the synthesis of appropriate metal complexes. The mono Schiff-base proligand H2tsc-difo was used to generate a nickel(II) complex [Ni(tsc-difo)PPh3] with triphenylphosphane as coligand leading to a complex with square-planar geometry at the nickel(II) center. As an example from the series of unsymmetric ditopic ligands we selected H2tsc-hydra due to its combination of a rigid and a flexible binding pocket. To explore its coordination behavior the homodinuclear zinc(II) complex [Zn2(tsc-hydra)(OAc)2] was synthesized. Interestingly two different coordination geometries were observed for the two zinc(II) ions indicating that the two distinctly different binding pockets of the ligand can enforce specific coordination modes which could be used to deliberately coordinate metal ions with corresponding geometric preferences for their coordination sphere. A specific potential can be attributed to complexes of the proligands as it should also be possible to utilize them for subsequent derivatization towards the generation of a second binding site with otherwise not accessible combinations of the underlying amine components.Acknowledgements
This work was in part supported by the Collaborative Research Centre ChemBioSys (CRC 1127 ChemBioSys) and funded by the Deutsche Forschungsgemeinschaft (DFG). We thank Stefan Schinkel for assistance in the assignment of the NMR data.References
- Handbook on Metalloproteins, ed. I. Bertini, A. Sigel and H. Sigel, Marcel Dekker, New York, 2001 Search PubMed.
- Handbook of Metalloproteins, ed. A. Messerschmidt, Wiley, New York, 2001-2011, vol. 1–5 Search PubMed.
- C. Belle, Eur. J. Inorg. Chem., 2003, 4137–4146 CrossRef CAS.
- J. J. H. Satcher, M. W. Droege, T. J. R. Weakley and R. T. Taylor, Inorg. Chem., 1995, 34, 3317–3328 CrossRef CAS.
- S. J. Lippard, Science, 1995, 268, 996–997 CAS.
- B. Zambelli, F. Musiani, S. Benini and S. Ciurli, Acc. Chem. Res., 2011, 44, 520–530 CrossRef CAS PubMed.
- T. Palzkill, Ann. N. Y. Acad. Sci., 2013, 1277, 91–104 CrossRef CAS PubMed.
- R. E. Stenkamp, Chem. Rev., 1994, 94, 715–726 CrossRef CAS.
- N. Mitić, S. J. Smith, A. Neves, L. Guddat, L. R. Gahan and G. Schenk, Chem. Rev., 2006, 106, 3338–3363 CrossRef PubMed.
- J. A. Tainer, E. D. Getzoff, J. S. Richardson and D. C. Richardson, Nature, 1983, 306, 284–287 CrossRef CAS PubMed.
- A. Roth and W. Plass, Angew. Chem., Int. Ed., 2008, 47, 7588–7591 CrossRef CAS PubMed.
- M. Carboni and J.-M. Latour, Coord. Chem. Rev., 2011, 255, 186–202 CrossRef CAS.
- S. Matsunaga and M. Shibasaki, Chem. Commun., 2014, 50, 1044–1057 RSC.
- I. Bratko and M. Gomez, Dalton Trans., 2013, 42, 10664–10681 RSC.
- D. G. H. Hetterscheid, S. H. Chikkali, B. de Bruin and J. N. H. Reek, ChemCatChem, 2013, 5, 2785–2793 CrossRef CAS.
- T. Hellmuth, S. Rieckhoff, M. Weiss, K. Dorst, W. Frey and R. Peters, ACS Catal., 2014, 4, 1850–1858 CrossRef CAS.
- B. Wu, J. C. Gallucci, J. R. Parquette and T. V. RajanBabu, Chem. Sci., 2014, 5, 1102–1117 RSC.
- S. W. S. Choy, M. J. Page, M. Bhadbhade and B. A. Messerle, Organometallics, 2013, 32, 4726–4729 CrossRef CAS.
- D. C. Powers and T. Ritter, Acc. Chem. Res., 2012, 45, 840–850 CrossRef CAS PubMed.
- M. Delferro and T. J. Marks, Chem. Rev., 2011, 111, 2450–2485 CrossRef CAS PubMed.
- J. P. McInnis, M. Delferro and T. J. Marks, Acc. Chem. Res., 2014, 47, 2545–2557 CrossRef CAS PubMed.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2004, 248, 1717–2128 CrossRef CAS.
- P. A. Vigato, V. Peruzzo and S. Tamburini, Coord. Chem. Rev., 2012, 256, 953–1114 CrossRef CAS.
- D. E. Fenton and H. Okawa, Chem. Ber./Recl., 1997, 130, 433–442 CrossRef CAS.
- A. L. Gavrilova and B. Bosnich, Chem. Rev., 2004, 104, 349–383 CrossRef CAS PubMed.
- P. J. Steel, Acc. Chem. Res., 2005, 38, 243–250 CrossRef CAS PubMed.
- M. Jarenmark, H. Carlsson and E. Nordlander, C. R. Chim., 2007, 10, 433–462 CrossRef CAS.
- P. A. Vigato and S. Tamburini, Coord. Chem. Rev., 2008, 252, 1871–1995 CrossRef CAS.
- K. E. Dalle and F. Meyer, Eur. J. Inorg. Chem., 2015, 3391–3405 CrossRef CAS.
- G. Belle, G. Gellon, C. Scheer and J.-L. Pierre, Tetrahedron Lett., 1994, 35, 7019–7022 CrossRef.
- M. Suzuki, S. Fujinami, T. Hibino, H. Hori, Y. Maeda, A. Uehara and M. Suzuki, Inorg. Chim. Acta, 1998, 283, 124–135 CrossRef CAS.
- T. Koga, H. Furutachi, T. Nakamura, N. Fukita, M. Ohba, K. Takahashi and H. Ōkawa, Inorg. Chem., 1998, 37, 989–996 CrossRef CAS.
- D. E. Fenton, Chem. Soc. Rev., 1999, 28, 159–168 RSC.
- R. Than, A. A. Feldmann and B. Krebs, Coord. Chem. Rev., 1999, 182, 211–241 CrossRef.
- H. Adams, L. R. Cummings, D. E. Fenton and P. E. McHugh, Inorg. Chem. Commun., 2003, 6, 19–22 CrossRef CAS.
- Y. Tachi, K. Aita, S. Teramae, F. Tani, Y. Naruta, S. Fukuzumi and S. Itoh, Inorg. Chem., 2004, 43, 4558–4560 CrossRef CAS PubMed.
- R. A. Peralta, A. Neves, A. J. Bortoluzzi, A. Casellato, A. dos Anjos, A. Greatti, F. R. Xavier and B. Szpoganicz, Inorg. Chem., 2005, 44, 7690–7692 CrossRef CAS PubMed.
- M. F. Anderlund, J. Högblom, W. Shi, P. Huang, L. Eriksson, H. Weihe, S. Styring, B. Åkermark, R. Lomoth and A. Magnuson, Eur. J. Inorg. Chem., 2006, 5033–5047 CrossRef CAS.
- Y.-W. Ren, J.-X. Lu, B.-W. Cai, D.-B. Shi, H.-F. Jiang, J. Chen, D. Zheng and B. Liu, Dalton Trans., 2011, 40, 1372–1381 RSC.
- M. Jarenmark, M. Haukka, S. Demeshko, F. Tuczek, L. Zuppiroli, F. Meyer and E. Nordlander, Inorg. Chem., 2011, 50, 3866–3887 CrossRef CAS PubMed.
- G. N. Ledesma and S. R. Signorella, Tetrahedron Lett., 2012, 53, 5699–5702 CrossRef CAS.
- L. J. Daumann, L. Marty, G. Schenk and L. R. Gahan, Dalton Trans., 2013, 42, 9574–9584 RSC.
- D. Wang, S. V. Lindeman and A. T. Fiedler, Inorg. Chim. Acta, 2014, 421, 559–567 CrossRef CAS.
- B. Das, H. Daver, M. Pyrkosz-Bulska, E. Persch, S. K. Barman, R. Mukherjee, E. Gumienna-Kontecka, M. Jarenmark, F. Himo and E. Nordlander, J. Inorg. Biochem., 2014, 132, 6–17 CrossRef CAS PubMed.
- K. C. Gupta and A. K. Sutar, Coord. Chem. Rev., 2008, 252, 1420–1450 CrossRef CAS.
- A. W. Kleij, Eur. J. Inorg. Chem., 2009, 193–205 CrossRef CAS.
- A. Roth, E. T. Spielberg and W. Plass, Inorg. Chem., 2007, 46, 4362–4364 CrossRef CAS PubMed.
- A. Roth, A. Buchholz, M. Rudolph, E. Schütze, E. Kothe and W. Plass, Chem.–Eur. J., 2008, 14, 1571–1583 CrossRef CAS PubMed.
- M. Ciaccia, S. Pilati, R. Cacciapaglia, L. Mandolini and S. D. Stefano, Org. Biomol. Chem., 2014, 12, 3282–3287 CAS.
- T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS.
- P. J. Jansson, P. C. Sharpe, P. V. Bernhardt and D. R. Richardson, J. Med. Chem., 2010, 53, 5759–5769 CrossRef CAS PubMed.
- D. R. M. Moreira, A. D. T. de Oliveira, P. A. T. de Moraes Gomes, C. A. de Simone, F. S. Villela, R. S. Ferreira, A. C. da Silva, T. A. R. dos Santos, M. C. A. B. de Castro, V. R. A. Pereira and A. C. L. Leite, Eur. J. Med. Chem., 2014, 75, 467–478 CrossRef CAS PubMed.
- L. Blau, R. F. Menegon, G. H. G. Trossini, J. V. D. Molino, D. G. Vital, R. M. B. Cicarelli, G. D. Passerini, P. L. Bosquesi and C. M. Chin, Eur. J. Med. Chem., 2013, 67, 142–151 CrossRef CAS PubMed.
- L. F. Lindoy, G. V. Meehan and N. Svenstrup, Synthesis, 1998, 1029–1032 CrossRef CAS.
- M. Schatz, M. Leibold, S. P. Foxon, M. Weitzer, F. W. Heinemann, F. Hampel, O. Walter and S. Schindler, Dalton Trans., 2003, 8, 1480–1487 RSC.
- R. W. W. Hooft, COLLECT, Data Collection Software, Nonius BV, Delft, Netherlands, 1998 Search PubMed.
- Z. Otwinowski and W. Minor, in Methods in Enzymology, Macromolecular Crystallography, Part A, ed. C. W. Carter and R. M. Sweet, Academic Press, New York, 1997, vol. 276, pp. 307–326 Search PubMed.
- SADABS 2.10, Bruker-AXS Inc., Madison, Wisconsin, USA, 2002 Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef CAS PubMed.
- L. Sobczyk, S. J. Grabowski and T. M. Krygowski, Chem. Rev., 2005, 105, 3513–3560 CrossRef CAS PubMed.
- P. Gilli, V. Bertolasi, V. Ferretti and G. Gilli, J. Am. Chem. Soc., 2000, 122, 10405–10417 CrossRef CAS.
- J. S. Casas, M. S. García-Tasende and J. Sordo, Coord. Chem. Rev., 2000, 209, 197–261 CrossRef CAS.
- T. S. Lobana, R. Sharma, G. Bawa and S. Khanna, Coord. Chem. Rev., 2009, 253, 977–1055 CrossRef CAS.
- R. Alonso, E. Bermejo, A. Custiñeiras, T. Pérez and R. Carballo, Z. Anorg. Allg. Chem., 1997, 623, 818–824 CrossRef CAS.
- H. Günther, NMR Spectroscopy, Wiley-VCH, Weinheim, 3rd edn, 2013 Search PubMed.
- K. G. Orrell, A. G. Osborne, J. O. Prince, V. Šik and D. K. Vellianitis, Eur. J. Inorg. Chem., 2000, 2000, 383–391 CrossRef.
- A. B. P. Lever, Inorganic Electronic Spectrocopy, Elsevier, Amsterdam, 2nd edn, 1984 Search PubMed.
- V. Philip, V. Suni, M. R. P. Kurup and M. Nethaji, Polyhedron, 2004, 23, 1225–1233 CrossRef CAS.
- N. T. Akinchan, R. Akinchan, U. J. Ibok, L. P. Battaglia, A. Bonamartini Corradi and P. Drozdzewski, J. Crystallogr. Spectrosc. Res., 1992, 22, 741–753 CrossRef CAS.
- T. S. Lobana, P. Kumari, G. Hundal and R. J. Butcher, Polyhedron, 2010, 29, 1130–1136 CrossRef CAS.
- R. Prabhakaran, P. Kalaivani, P. Poornima, F. Dallemer, G. Paramaguru, V. V. Padma, R. Renganathan, R. Huang and K. Natarajan, Dalton Trans., 2012, 41, 9323–9336 RSC.
- O. Kühl, in The Range of Chemical Shifts, Coupling Constants, and What Influences Each, ed. O. Kühl, Springer-Verlag, Berlin Heidelberg, 2008, ch. 2, pp. 7–23 Search PubMed.
- L. Pazderski, Annu. Rep. NMR Spectrosc., 2013, 80, 33–179 CrossRef CAS.
- T. Steiner and G. Koellner, J. Mol. Biol., 2001, 305, 535–557 CrossRef CAS PubMed.
- O. Takahashi, Y. Kohno and M. Nishio, Chem. Rev., 2010, 110, 6049–6076 CrossRef CAS PubMed.
- W. Plass, Z. Anorg. Allg. Chem., 1997, 623, 461–477 CrossRef CAS.
- W. Plass, Z. Anorg. Allg. Chem., 1994, 620, 1635–1644 CrossRef CAS.
- J. B. Mandel, C. Maricondi and B. E. Douglas, Inorg. Chem., 1988, 27, 2990–2996 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC numbers 1043774 ([Zn2(tsc-hydra)(OAc)2]) and 1048856 ([Ni(tsc-difo)PPh3]). For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra16870a |
| This journal is © The Royal Society of Chemistry 2016 |