
The synthesis and comparison of poly(methacrylic acid)–poly(ε-caprolactone) block copolymers with and without symmetrical disulfide linkages in the center for enhanced cellular uptake†
Abstract
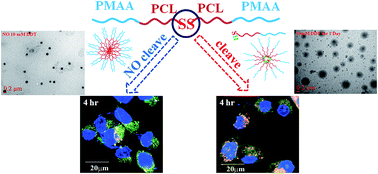
A self-assembled poly(methacrylic acid)–poly(ε-caprolactone) block copolymer with a disulfide linkage, PMAA-b-PCL-SS-PCL-b-PMAA (S-PCL-PMAA)2, was synthesized for enhanced cellular uptake due to a reduction response to glutathione (GSH) and pH-sensitive characteristics. For comparison, a reduction-insensitive PMAA-b-PCL-CC-PCL-b-PMAA (C-PCL-PMAA)2, using a carbon–carbon linkage as a symmetrical center was also synthesized. These block copolymers were synthesized via the combination of ring opening polymerization (ROP) and atom transfer radical polymerization (ATRP) followed by hydrolysis. The similar number of MAA repeating units was controlled in the copolymers containing either disulfide or carbon–carbon linkages. Copolymers could self-assemble into core–shell micelles in an aqueous solution owing to amphiphilicity. The molecular weight of (S-PCL-PMAA)2 increases linearly with reaction time at both reaction temperatures of 40 and 80 °C. Critical micelle concentrations range within 3.09 × 10−3 to 6.31 × 10−3 mg mL−1 at pH 5 and 3.16 × 10−2 to 3.98 × 10−2 mg mL−1 at pH 8. The average hydrodynamic diameters of micelles are ∼200 nm. The cellular uptake of (S-PCL-PMAA)2 increases with incubation time and is higher in the medium with GSH than without in CRL-5802 cells. In contrast, the cellular uptake of (C-PCL-PMAA)2 is insensitive to the presence of GSH. The higher internalization of the micelle containing disulfides in the presence of GSH is attributable to all three pinocytosis pathways involved, including macropinocytosis-, caveolae-, clathrin-mediated endocytosis, but in the absence of GSH clathrin-mediated endocytosis is only involved. The nascent (S-PCL-PMAA)2 is non-cytotoxic to four cell lines. However, the paclitaxel-encapsulated (S-PCL-PMAA)2 shows slightly higher cell-killing ability than free paclitaxel against CRL-5802 cells. Thus, the GSH-responsive (S-PCL-PMAA)2 is a potential drug delivery system.
 Please wait while we load your content...
Please wait while we load your content...

