
Syntheses, structures and magnetic properties of symmetrical Schiff-base supported dinuclear Ln(iii) compounds†
Abstract
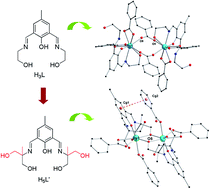
Three dinuclear Ln(III) compounds, [Ln2(H3L)2(PhCOO)6] (Ln = Sm (1), Dy (2)) and [Dy2(H4L′)2(PhCOO)4]·2CH3CN (3) have been synthesized and structurally and magnetically characterized. Structure analyses revealed distinct structural features in compounds with different symmetrical arms of Schiff-base ligands. With the symmetrical two-arm Schiff-base ligand H3L, dinuclear Ln(III) compounds 1 and 2 were synthesized, where nine-coordinated Ln(III) ions were bridged by two syn–syn η1:η1-µ2-benzoate groups. When using symmetrical Schiff-base ligand H5L′ with four arms, the dinuclear Dy(III) compound 3 was prepared, in which eight-coordinated Dy ions were bridged by two alkoxido oxygen atoms from additional arms of H5L′. Direct-current (DC) magnetic susceptibility studies revealed that the Dy(III) ions were very weakly coupled in dysprosium compounds. Alternating-current (AC) magnetic susceptibility studies for compounds 2 and 3 indicated that field-induced slow relaxation phenomenon occured for both compounds. Furthermore, two relaxation phases under the optimal field appeared in compound 3, which are probably associated with the existence of the anisotropic Dy(III) ion for the slow relaxation phase (SR) and significant quantum tunneling for the fast relaxation phase (FR).
 Please wait while we load your content...
Please wait while we load your content...

