
A series of tetraphenylethene-based benzimidazoles: syntheses, structures, aggregation-induced emission and reversible mechanochromism†
Tengfei Zhanga,
Ran Zhanga,
Zhaoming Zhangb and
Zhonghai Ni*a
aSchool of Chemical Engineering and Technology, China University of Mining and Technology, Xuzhou 221116, Jiangsu Province, P. R. China. E-mail: nizhonghai@cumt.edu.cn
bInstitute for Catalysis, Hokkaido University, Sapporo, 001-0021, Japan
First published on 17th August 2016
Abstract
Four tetraphenylethene-based benzimidazoles 5,6-R-2-(4-tetraphenyletheneyl)-1H-benzo[d]imidazoles (R = H (2a), Cl (2b), Br (2c) and CH3 (2d)) were conveniently synthesized in high yields by the cyclization reaction of 4-tetraphenylenthenealdehyde with phenylenediamines. These compounds were structurally characterized and their properties were analyzed by spectroscopy, electrochemistry, X-ray diffraction, thermal stability and theoretical studies. Single crystal X-ray diffraction analyses indicate that their molecular structures in the aggregated state are highly twisted conformations and there exist obvious cavities and/or interface gaps in their crystal packing structures. These four compounds exhibit typical aggregation-induced emission (AIE) properties with relatively high absolute fluorescence quantum yields (30.6–67.5%). These compounds also exhibit reversible mechanochromism behaviors with emission changes between light blue and yellow-green, which are due to the reversible morphological transformation between unconsolidated crystalline state and amorphous state. They are thermally stable with decomposition temperatures above 340 °C.
Introduction
Recently, organic photo-active materials are being investigated widely because of their enormous application value in organic light emitting diodes, sensors and fluorescent probes.1–8 Generally, enlarging the molecular π-conjugation structure is one of the most effective strategies for obtaining excellent organic photo-active materials. However, the simple enlargement of the π-conjugated electronic structure usually leads to one very detrimental result that is aggregation-caused quenching (ACQ),9,10 which significantly limits the application of organic photo-active materials in their solid state. Fortunately, Tang et al.11 found the phenomenon of aggregation-induced emission (AIE). Tetraphenylethene (TPE), as the most typical representative AIE-active compound, has attracted much research attention because it benefits from the advantages of efficient solid-state emission, facile synthesis and easy functionalization.12,13Mechanochromism is another intriguing phenomenon of photo-active materials that the emission of compounds can be changed under the outer mechanical pressure in the solid state. To achieve vivid emission color change under external pressure, photo-active compounds should exhibit strong emission in solid state. Furthermore, TPE derivatives exhibit mechanochromism by simply changing their molecular packing mode.14–16 Therefore, the design and synthesis of organic compounds using TPE as the precursor should be one convenient strategy to obtain AIE-active organic photoelectric materials with mechanochromism at the same time.
Based on the above considerations, we designed and synthesized a series of TPE-based benzimidazoles, in which benzimidazole segment not only increases the extent of π-electron delocalization of the system, but also may bring the change of intermolecular interactions especially the intermolecular hydrogen bonds under outer pressure. The different peripheral substituent groups including H, CH3, Cl and Br can also affect their photophysical properties. Expectedly, all the four compounds exhibit bifunctional AIE and reversible mechanochromic property, furthermore, the change of the substituents has a great effect on emission efficiency. Herein, this paper report the syntheses, structures, AIE, reversible mechanochromism, thermal stability and electrochemical properties as well the theoretical studies of the new series of TPE-based benzimidazoles.
Results and discussion
Synthesis and characterization
The synthetic procedures of the four compounds are illustrated in Scheme 1 using simple TPE derivative 4-Br-tetraphenylethene as the starting material. Firstly, 4-Br-tetraphenylethene was transformed into the key intermediate 4-tetraphenylenthenealdehyde (1) according to the conventional method as literature reported.17 Then, the four compounds 2a–2d were synthesized by the typical cyclization reaction of the intermediate 1 with phenylenediamines in high yields.18,19 These compounds were purified by column chromatography followed by recrystallization. They were full structurally characterized by 1H-NMR, 13C-NMR, single crystal X-ray diffraction and MALDI TOF/TOF mass spectrometer techniques. Generally, four new TPE-based benzimidazoles have been synthesized conveniently using simple TPE derivative as the precursor. | ||
| Scheme 1 Procedures for the syntheses of compounds 2a–2d. | ||
Crystal structures
X-ray single diffraction analyses of compounds 2a–2d agree well with the expected structures as displayed in Fig. 1. Similar to the reported TPE-based compounds,20–25 the four compounds show highly twisted paddle-shaped conformations. The benzimidazole segments with different substituents are attached to the peripheral position of TPE unit in compounds 2a–2d. The benzimidazole segment and the adjacent benzene ring of TPE unit are nearly planar with the dihedral angles in the small range of 3.03–5.72° for compounds 2b–2d. But for compound 2a, there are two different dihedral angles (7.12° and 24.88°) between benzimidazole segment and the adjacent benzene ring of TPE unit because there are two independent molecular units for compound 2a. | ||
| Fig. 1 Crystal structures of compounds 2a–2d. Solvent molecules and hydrogen atoms bonded to carbon atoms were omitted for clarity. | ||
There exist very different hydrogen bonds26–28 in the crystal structures of the four compounds, yielding different supramolecular structures as shown in Fig. 2. For compound 2a, the molecular units of 2a are alternating crossly linked together by N–H⋯N hydrogen bonds between adjacent benzimidazole segments, forming X-shaped one-dimensional (1D) supramolecular structure. For compound 2b, molecular units of 2b are linked together by N–H⋯O and O–H⋯N hydrogen bonds between free water molecules and benzimidazole segments, giving zigzag-shaped 1D chain-like supramolecular structure. Although compound 2d also forms 1D chain-like supramolecular structure through N–H⋯O and O–H⋯N hydrogen bonds involving free methanol molecules and benzimidazole segments, all the benzimidazole segments in the chain are parallel to each other. However, compound 2c only forms supramolecular dimeric structure in which two units of 2c are connected by one free water molecule through O–H⋯N hydrogen bonds.
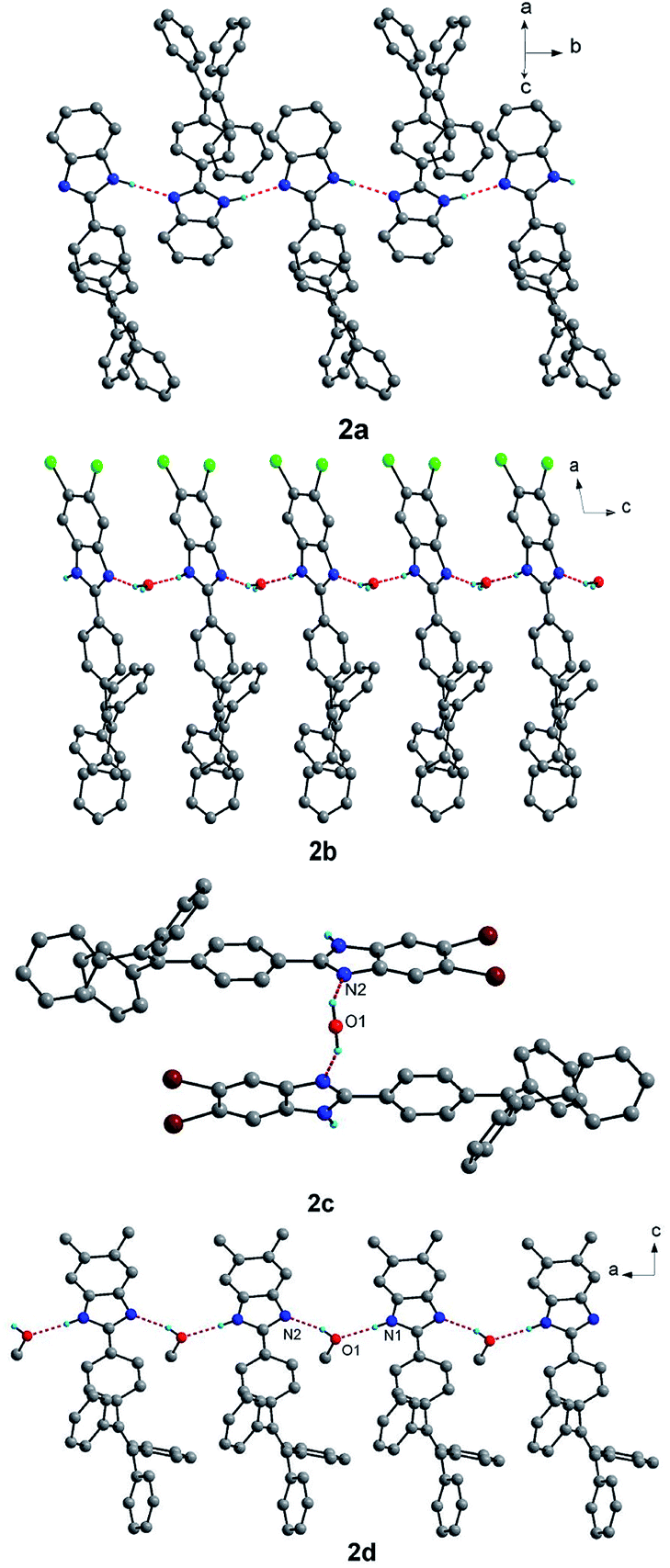 | ||
| Fig. 2 Supramolecular structures of compounds 2a–2d linked by hydrogen bonds. Some solvent molecules and hydrogen atoms bonded to carbon atoms were omitted for clarity. | ||
There also exist affluent intermolecular C–H⋯π interactions in the crystal structures of the four compounds and intermolecular π–π interactions for 2b and 2d (see Fig. S1–S4†), which link the above hydrogen bond-based supramolecular structures to form three-dimensional (3D) supramolecular networks. There are not obvious intermolecular π–π interactions in the crystal structures of compounds 2a and 2c. However, for compound 2b, there exist relatively strong slipped intermolecular π–π interactions between two adjacent benzimidazole segments (3.437(4) Å for the shortest distance of atoms) and between two adjacent benzene rings (3.692(4) Å for the shortest distance of atoms) of TPE units. For compound 2d, there also exist slipped intermolecular π–π stacking interactions in two adjacent benzimidazole segments. In addition, there exist relatively significant cavities and/or interface gaps which accommodate solvent guest molecules in the crystal packing structures of the four compounds (Fig. S5†). Due to existence of weak intermolecular interactions, obvious cavities and/or interfaces in the four compounds, the crystal packing interactions are readily destroyed by external pressure, which may result in mechanochromism.
Optical properties
The UV-visible spectra of the four compounds are demonstrated in Fig. 3 and the data are summarized in Table 1. Compounds 2a–2d exhibit similar absorption behaviors at absorption band between 230 and 400 nm and absorption maxima are observed at 337 for 2a, 343 for 2b, 345 for 2c and 345 nm for 2d, respectively. Compared to the absorption maxima of compound 2a, the other three compounds show a bathochromic shift, which can be due to the n–π conjugation (2b and 2c) of halogen with benzimidazole unit and hyper conjugation (2d) of methyl with benzimidazole unit. The results of photoluminescence (PL) spectra indicate that all the four compounds display weak emissions in tetrahydrofuran (THF). The relative fluorescent quantum yields 2a–2d using 9,10-diphenylanthracene (ΦF = 90% in cyclohexane) as the standard have been estimated with the small values in the range of 0.16–0.22%. The non-radiative decay of the excited states of compounds 2a–2d resulted from intramolecular rotations of free benzene rings and benzimidazole units is responsible for their low fluorescence quantum yields in dilute THF solution. | ||
| Fig. 3 (a) UV-visible spectra of the dilute solutions of compounds 2a–2d in THF (concentration: 50 μM). (b) Normalized emission spectra of the compounds 2a–2d recorded in solid. | ||
| Compounds | λabsa (nm) | ΦFb (%) | λpristine (nm) | λgrinded (nm) | λfumed (nm) | Δλc (nm) | λemd (nm) |
|---|---|---|---|---|---|---|---|
| a Measured in tetrahydrofuran.b Measured in pristine solid.c Δλ = λgrinded − λpristine.d Measured in THF/water mixtures (fw = 90%). | |||||||
| 2a | 337 | 67.5 | 465 | 492 | 467 | 27 | 486 |
| 2b | 343 | 30.6 | 467 | 493 | 468 | 26 | 487 |
| 2c | 345 | 45.6 | 461 | 487 | 463 | 26 | 487 |
| 2d | 345 | 38.2 | 461 | 485 | 462 | 24 | 487 |
Unlike the PL spectra in THF solution, intense emissions have been obtained when the samples are in solid forms. As shown in Fig. 3b, the corresponding emission peaks of compounds 2a–2d appear at 465, 467, 461 and 461 nm, respectively. The measurements of the fluorescence quantum yields reveal that all of the compounds are strong solid state emitters with the ΦF values of 67.5% for 2a, 30.6% for 2b, 45.6% for 2c and 38.2% for 2d, respectively. The restriction of RIR29–31 mechanism may account for the different emission behaviors between their solution and solid state. In contrast to their active free rotation in solution along Caryl–Caryl σ bond, the rotation of benzene rings and benzimidazole units are restricted effectively. As a result, the non-radiative channel is blocked, which enhances the emission in solid state.32,33 The ΦF values of compounds 2b and 2d are lower than those of 2a and 2c, which may be attributed to the existence of relatively stronger π–π interactions in the two compounds as shown in their crystal structures. Besides, the ΦF values of compounds 2b and 2c are lower than compound 2a, which is due to heavy atom effect that enhances the rate of singlet-to-triplet intersystem crossing (ISC).34–36
AIE properties
In order to further investigate the AIE effects of the four compounds, the AIE properties of 2a–2d were studied by PL spectra in THF/water mixtures. Compounds 2a–2d are highly soluble in THF and insoluble in water, with increasing the water fraction in THF/water mixtures, invisible particulates are formed, leading to the change in PL spectra (Fig. 4a and S6†). At the beginning, the emissions of compounds 2a–2d are weakly fluorescent in pure THF solution. When the water fraction is increased, these weak emissions keep constantly until about the water fraction up to 75% in THF/water mixture. Then, with further increasing the water fraction, all the emissions of compounds 2a–2d sharply increase until the measured water fraction of 90%. The above emission tendencies clearly exhibit that the four compounds are typical AIE-active compounds. The faint fluorescence emission in THF solution is enhanced in the aggregated suspension (90% water fraction) by 238-, 36-, 156- and 116-fold for compounds 2a–2d, respectively (Fig. 4b). The emission enhancement ratio of compounds 2b and 2d are lower than those of compounds 2a and 2c, which can be due to the existence of relatively strong intermolecular π–π interactions in compounds 2b and 2d. The emission peaks of the four compounds in aggregation state are close to that measured in grinded solid state (Table 1), indicating the packing mode of aggregative state is similar to that of grinded amorphous solid state. This phenomenon indicates that the molecules quickly assemble in an irregularity fashion, forming random style aggregates abruptly when the water fraction is above 75%. | ||
| Fig. 4 (a) Fluorescence spectra of 2a in THF/water mixtures with different water fractions, inset in panel (a): photos of compounds 2a in THF/water mixtures (fw = 0 and 90%) under 365 nm UV illumination. (b) Plots of (I/I0) values versus the compositions of the THF/H2O mixtures of compounds 2a–2d, where I0 is the PL intensity in pure THF solution. Luminogen concentration: 50 μM; excitation wavelength: 365 nm; intensity calculated at λmax. | ||
Theoretical calculation and electrochemical studies
To further understand the correlation between structure and the physical properties, we performed quantum mechanical computations by density functional theory (DFT) calculations using the Gaussian 09W software.37 The DFT calculations were performed at the B3LYP/6-31G(d,p) level of theory. The contours of the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) of 2a–2d are shown in Fig. S7.† The HOMO and LUMO of four compounds are majorly distributed on the whole molecules, indicating that the absorption and emission behaviors are related to the π–π* transitions and exciton decays of the whole molecules. The theoretical band gap for 2a–2d is 3.74 eV, 3.67 eV, 3.67 eV and 3.68 eV, respectively (Fig. S8†). The theoretical band gaps of compounds 2a–2d are largely identical but with minor differences. Furthermore, the general order of band gap is in good agreement with that the maximum absorption wavelength. The low band-gap suggests a bathochromic shift in the absorption spectra.The electrochemical properties were investigated by cyclic voltammetry (CV) measurements of the four compounds in anhydrous tetrahydrofuran with the concentration of 1.0 × 10−4 M. The corresponding data are summarized in Table 2. On the basis of the onset potentials of oxidation (Eoxonset), the HOMO energy levels were calculated with the values of 5.83, 5.83, 5.81 and 5.79 eV respectively using the equation HOMO = −eEonset − 4.19 eV. These results are slightly lower than that obtained by theoretical calculation. The band gaps were derived from the absorption edge in the absorption spectra with the values of 3.19 eV for 2a, 3.14 eV for 2b, 3.13 eV for 2c and 3.12 eV for 2d. Though the calculated values are somewhat higher than the values estimated by CV and have little difference each other, the trend is similar with the order of maximum absorption wavelength. This trend can nicely explain the bathochromic shift in the absorption of compounds 2b–2d compared with 2a. The LUMO values of samples can be estimated by subtraction of the optical band gap energies from the HOMO energy levels with the values of 2.64, 2.69, 2.68 and 2.67 eV for compounds 2a–2d, respectively.
| Compounds | Eoxonset (V) | HOMO (eV) | LUMO (eV) | Eg (eV) | Tg/Td (°C) | |||
|---|---|---|---|---|---|---|---|---|
| Exptla | Calc. | Exptl | Calc. | Exptlb | Calc. | |||
| a HOMO levels were determined using the following equations: HOMO = −e(Eonset − 0.61 V) − 4.8 eV, where the value 0.61 V is for FOC versus SCE electrode.b Estimated from the onset of the absorption spectra: 1240/λonset. | ||||||||
| 2a | 1.64 | −5.83 | −5.29 | −2.64 | −1.55 | 3.19 | 3.74 | 128/350 |
| 2b | 1.64 | −5.83 | −5.47 | −2.69 | −1.80 | 3.14 | 3.67 | 132/355 |
| 2c | 1.62 | −5.81 | −5.47 | −2.68 | −1.80 | 3.13 | 3.67 | 139/375 |
| 2d | 1.60 | −5.79 | −5.18 | −2.67 | −1.50 | 3.12 | 3.68 | 142/340 |
Thermal studies
The thermal properties of compounds 2a–2d were studied by thermo-gravimetric analysis (TGA) and differential scanning calorimetry (DSC) analyses as shown in Fig. 5. All of the compounds show high thermal decomposition (Td) temperatures with the values of 350, 355, 375 and 340 °C for compounds 2a–2d, respectively. The DSC results of compounds 2a–2d show relatively high glass transition temperatures with Tg values of 128, 132, 139 and 142 °C, respectively. As shown in Fig. S9,† the melting peaks of compounds 2a–2d at first heating scan are 269 °C, 282 °C, 262 °C and 323 °C, respectively. During the second heating scan, except compound 2c, the other compounds show a cold-crystallization peak before melting peak. In addition, compound 2a shows a large cold-crystallization peak. For compounds 2b and 2d, the cold-crystallization peaks become very weak. However, the cold-crystallization peak disappears for compounds 2c. The results indicate that crystallinity property of these compounds is related with the peripheral substituted groups and the heavy atoms such as Br do not facilitate the transformation from amorphous state to crystalline state during the second non-isothermal heating process. | ||
| Fig. 5 (a) Thermogravimetric analysis and (b) DSC curves (second heating scan) of compounds 2a–2d recorded under nitrogen at a heating rate of 10 °C min−1. | ||
Mechanochromic properties
The mechanochromic properties of compounds 2a–2d were studied by solid state emission spectroscopy and the data are summarized in Table 1. Compounds 2a–2d show sky blue emission with wavelengths ranging from 450 nm to 467 nm in their solid crystal state. Upon grinding by a spatula or a pestle, the sky blue emitting solids can convert to yellowish green emitting solids and the emission wavelength is red-shifted to a range from 485 to 493 nm (Fig. 6a and S10†). The compounds 2a–2d show grinding-induced spectral red shift (Δλ) of 27 nm, 26 nm, 26 nm and 24 nm, respectively. It is noteworthy that this mechanochromic effect of the four compounds is reversible. After the ground samples of these four compounds are heated or fumed with dichloromethane vapor, the PL wavelengths of ground samples are blue-shifted to those of their original crystal state completely. This mechanochromic conversion can be repeated between light blue and yellowish green emissions (6 times) as these stimuli do not cause any chemical change (Fig. S11†).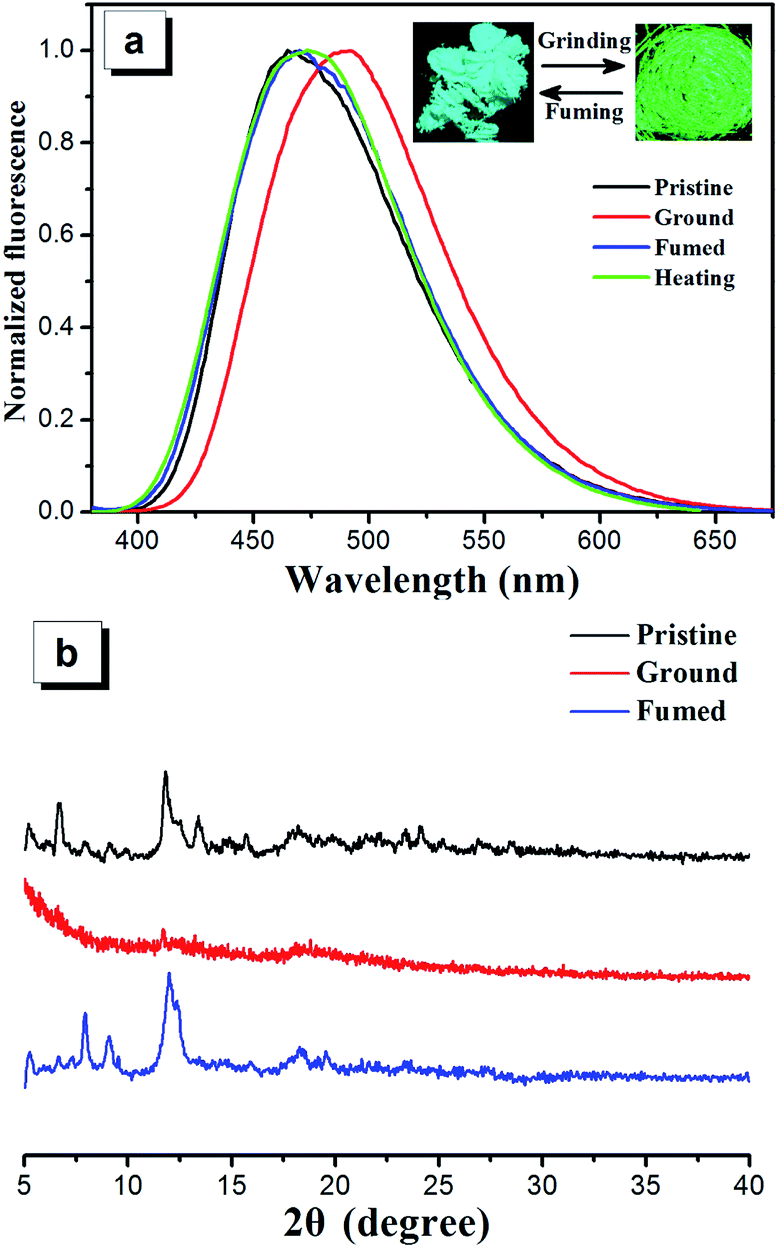 | ||
| Fig. 6 (a) Emission spectra of 2a as pristine, grinded, fumed and heated solids, inset in panel (a): photos of compounds 2a in pristine (left) and ground (right) forms under 365 nm UV illumination. (b) Powder X-ray diffraction patterns of 2a in pristine, ground and fumed forms. | ||
The single crystal structures of the compounds 2a–2d provide more insights into the AIE and mechanochromic mechanisms because such photo-active behaviors are usually related with their packing patterns and the stacking interactions in the solid state. According to the crystal structures, the four compounds adopt highly twisted conformations, leading to unconsolidated stacking. Moreover, there exist obvious cavities and/or interface gaps which contain solvent molecules in the crystal structures of compounds 2a–2d. Therefore, the unconsolidated structures are readily destroyed, making the formation of relatively tight planar conformations under external mechanical pressure, which increases the effective intermolecular and intramolecular conjugation effects and results in the red-shift of emissions.38 However, after heated or fumed with dichloromethane vapor, the molecules can reassemble into their original unconsolidated stacking constructions, which is responsible for the emitting recovery to their original crystal state of the four compounds.
In order to further understand the mechanism of mechanochromism, different solid states of compounds 2a–2d were studied by powder X-ray diffraction (PXRD) (Fig. 6b and S10†). The diffraction patterns for pristine samples of 2a–2d show representative sharp peaks, which is the characteristic properties of the crystalline state. The pristine samples of 2a–2d upon grinding show very weak diffraction signal intensity or a broad diffuse band, indicating that the packing modes change from crystalline state to amorphous state. The ground samples can restore the sharp peaks after heating or fuming with dichloromethane and these peaks are consistent with those of corresponding original crystals, demonstrating recoverability of crystal morphology from amorphous state to crystalline state. Therefore, the reversible mechanochromism of compounds 2a–2d can be attributed to the reversible morphological transformation between crystalline state and amorphous state.39–43
Experimental
Materials and instrumentation
THF was purified by distillation from sodium and benzophenone ketyl under nitrogen condition prior to use. All other reagents and solvents were purchased commercially (AR grade) and used without further purification unless otherwise noted. The THF/water mixtures with different water fractions were prepared by slowly adding distilled water into the THF solution of samples under ultrasound at room temperature. The single crystals suitable for X-ray diffraction analysis were obtained by the slow evaporation of acetonitrile/water mixture (2a–2c) and methanol/water (2d) mixture. 1H-NMR and 13C-NMR spectra were collected on a Bruker-400 MHz or Bruker-600 MHz spectrometer in CDCl3 or DMSO solutions with TMS as an internal standard. Mass spectra were obtained on a Bruker Ultraflextreme MALDI TOF/TOF mass spectrometer and a Jms-Q1000GC MKIIUltra Quad GC/MS. The ΦF values of the pristine solid were determined by FM-4P-TCSPC Transient State Fluorescence Spectrometer using an integrating sphere. UV-vis spectra were recorded on Shimadzu UV-3600 with a UV-VIS-NIR spectrophotometer. Emission spectra were performed by a HITACHI fluorescence spectrometer (F-4600). Crystal data of compounds were selected on a Bruker APEX II CCDC diffractometer with graphite monochromated Mo-Kα radiation (λ = 0.71073 Å) at 293 K using the ω-scan technique. The structure was solved by direct methods with the SHELXS-97 (ref. 44) computer program, and refined by full-matrix least-squares methods (SHELXL-97) on F2. The ground state geometries of all molecules were fully optimized using density functional theory (DFT) at the B3LYP/6-31G(d,p) level, as implemented in Gaussian 09W software package. PXRD measurements were performed by using a Bruker X-ray diffractometer (D8 Advance, Germany) with an X-ray source of Cu-Kα (λ = 0.15418 Å) at 40 kV and 40 mA at a scan rate of 4°(2θ) min−1. Cyclic voltammetry experiments were performed with a CHI660A electrochemical work station. All measurements were carried out at room temperature with a conventional three-electrode configuration consisting of a glassy carbon working electrode, a platinum auxiliary electrode and a calomel reference electrode. The solvent in all experiments was dry tetrahydrofuran and the supporting electrolyte was 0.1 M tetrabutylammonium hexafluorophosphate. The glass-transition temperatures (Tg) of the compounds were determined with differential scanning calorimetry (DSC) under a nitrogen atmosphere by using a DSC6000 (PerkinElmer). Samples were heated to 300 °C at a rate of 10 °C min−1 and cooled at 20 °C min−1 then heated again under the same heating conditions as used in the initial heating process. Decomposition temperatures (Td) were determined with thermogravimetric analysis (TGA) under a nitrogen atmosphere by using a STA 449 F5 (NETZSCH). Samples were heated to 600 °C at a rate of 10 °C min−1.Synthesis
Compound 2a was obtained as a pale yellow powder solid. Yield: 0.30 g, 67.0%. 1H-NMR (600 MHz, d6-DMSO) δ (ppm): 12.81 (s, 1H), 7.93 (d, J = 8.4 Hz, 1H), 7.63 (d, J = 7.8 Hz, 1H), 7.49 (d, J = 7.8 Hz, 1H), 7.24–7.09 (m, 13H), 7.06–6.98 (m, 6H). MALDI TOF-MS: m/z 448.296 [M]+.
Compound 2b was obtained as a white powder solid. Yield: 0.42 g, 81.4%. 1H-NMR (400 MHz, d6-DMSO) δ (ppm): 13.17 (s, 1H), 7.93 (d, J = 8.5 Hz, 2H), 7.21–7.11 (m, 12H), 7.07–6.97 (m, 7H). 13C-NMR (101 MHz, d6-DMSO) δ (ppm): 153.97 (s), 146.01 (s), 143.38 (s), 142.05 (s), 140.28 (s), 131.81 (s), 131.14 (d, J = 10.7 Hz), 128.40 (d, J = 11.8 Hz), 127.26 (s), 126.69 (s). MALDI TOF-MS: m/z 516.210 [M]+.
Compound 2c was obtained as a pale yellow powder solid. Yield: 0.47 g, 77.8%. 1H-NMR (400 MHz, d6-DMSO) δ (ppm): 13.16 (s, 1H), 7.92 (d, J = 8.5 Hz, 4H), 7.28–7.08 (m, 11H), 7.09–6.91 (m, 6H). 13C-NMR (101 MHz, d6-DMSO) δ (ppm): 153.75 (s), 146.04 (s), 143.28 (d, J = 19.1 Hz), 142.05 (s), 140.28 (s), 131.81 (s), 131.13 (d, J = 7.8 Hz), 128.39 (d, J = 11.8 Hz), 127.67 (s), 127.32 (s), 126.74 (s). MALDI TOF-MS: m/z 606.150 [M]+.
Compound 2d was obtained as a pale yellow powder solid. Yield: 0.35 g, 73.5%. 1H-NMR (400 MHz, d6-DMSO) δ (ppm): 12.54 (s, 1H), 7.88 (d, J = 8.5 Hz, 2H), 7.31 (s, 1H), 7.14 (ddd, J = 20.8 Hz, 15.0 Hz, 7.8 Hz, 12H), 7.06–6.98 (m, 6H). 13C-NMR (101 MHz, d6-DMSO) δ (ppm): 150.45 (s), 144.85 (s), 143.42 (d, J = 19.3 Hz), 141.71 (s), 140.47 (s), 131.63 (s), 131.41–130.95 (m), 128.88 (s), 128.37 (d, J = 9.6 Hz), 127.23 (s), 126.11 (s), 20.49 (s). MALDI TOF-MS: m/z 477.199 [M + 1]+.
Conclusions
In conclusion, we have designed and synthesized a new series of tetraphenylethene-based benzimidazoles 2a–2d. All the compounds are weakly emissive in solution but become strong emitters in aggregation state, demonstrating that they are typical AIE-active compounds with relatively excellent absolute fluorescence quantum yields (30.6–67.5%). Their single crystal structures demonstrate unconsolidated molecular stacking structures and there are affluent cavities and/or interface gaps in crystal structures of the four compounds. These compounds exhibit reversible mechanochromism behaviors with emission changes between light blue and yellow-green, the PXRD results reveal that the reversible morphological transformation of the twisted crystalline state to the planar amorphous state is the main cause of mechanochromism in the compounds 2a–2d. Thermal studies show that the four compounds exhibit a relatively high thermal stability and glass transition temperature. The work further show it is still one of the most effective strategies using the known TPE and its simple derivatives as the starting materials for novel photo-active especially AIE-active materials and OLED application of new AIE compounds are under investigation in our lab.Acknowledgements
This work was supported by the Fundamental Research Funds for the Central Universities (2015XKZD08) and the Priority Academic Program Development of Jiangsu Higher Education Institutions.Notes and references
- S. R. Forrest, Org. Electron., 2003, 4, 45 CrossRef.
- O. S. Wolfbeis, J. Mater. Chem., 2005, 15, 2657 RSC.
- T. P. I. Saragi, T. Spehr, A. Siebert, T. Fuhrmann-Lieker and J. Salbeck, Chem. Rev., 2007, 107, 1011 CrossRef CAS PubMed.
- F. Cicoira and C. Santato, Adv. Funct. Mater., 2007, 17, 3421 CrossRef CAS.
- L. Yuan, W. Y. Lin, K. B. Zheng, W. L. He and M. W. Huang, Chem. Soc. Rev., 2013, 42, 622 RSC.
- H. Xu, R. F. Chen, Q. Sun, W. Y. Lai, Q. Q. Su, W. Huang and X. G. Liu, Chem. Soc. Rev., 2014, 43, 3259 RSC.
- R. Zhang, Y. Zhao, G. L. Li, D. S. Yang and Z. H. Ni, RSC Adv., 2016, 6, 9037 RSC.
- R. Zhang, Y. Zhao, T. F. Zhang, L. Xu and Z. H. Ni, Dyes Pigm., 2016, 130, 106 CrossRef CAS.
- H. J. Tracy, J. L. Mullin, W. T. Klooster, J. A. Martin, J. Haug, S. Wallace, I. Rudloe and K. Watts, Inorg. Chem., 2005, 44, 2003 CrossRef CAS PubMed.
- S. Jayanty and T. P. Radhakrishnan, Chem.–Eur. J., 2004, 10, 791 CrossRef CAS PubMed.
- Y. N. Hong, J. W. Y. Lam and B. Z. Tang, Chem. Commun., 2009, 4332 RSC.
- L. Chen, Y. B. Jiang, H. Nie, R. R. Hu, H. S. Kwok, F. Huang, A. J. Qin, Z. J. Zhao and B. Z. Tang, ACS Appl. Mater. Interfaces, 2014, 6, 17215 CAS.
- Y. N. Hong, M. H. Uble, J. W. Y. Lam, Z. Li, K. K. Sin, Y. Q. Dong, H. Tong, J. Z. Liu, A. J. Qin, R. Renneberg and B. Z. Tang, Chem.–Eur. J., 2008, 14, 6428 CrossRef CAS PubMed.
- B. J. Xu, M. Y. Xie, J. J. He, B. Xu, Z. G. Chi, W. J. Tian, L. Jiang, F. L. Zhao, S. W. Liu, Y. Zhang, Z. Z. Xu and J. R. Xu, Chem. Commun., 2013, 49, 273–275 RSC.
- W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H.-Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837–2843 CrossRef CAS PubMed.
- F. F. Han, R. Zhang, Z. M. Zhang, J. G. Su and Z. H. Ni, RSC Adv., 2016, 6, 68178 RSC.
- R. R. Hu, J. L. Maldonado, M. Rodriguez, C. M. Deng, C. K. W. Jim, J. W. Y. Lam, M. M. F. Yuen, G. Ramos-Ortiz and B. Z. Tang, J. Mater. Chem., 2012, 22, 232 RSC.
- M. P. Singh, S. Sasmal, W. Lu and M. N. Chatterjee, Synthesis, 2000, 10, 1380 CrossRef.
- K. Bahrami, M. M. Khodaei and F. Naali, J. Org. Chem., 2008, 73, 6835 CrossRef CAS PubMed.
- Z. J. Zhao, S. M. Chen, J. W. Y. Lam, Z. M. Wang, P. Lu, F. Mahtab, H. H. Y. Sung, I. D. Williams, Y. G. Ma, H. S. Kwok and B. Z. Tang, J. Mater. Chem., 2011, 21, 7210 RSC.
- N. Zhao, Z. Y. Yang, J. W. Y. Lam, H. H. Y. Sung, N. Xie, S. J. Chen, H. M. Su, M. Gao, I. D. Williams, K. S. Wong and B. Z. Tang, Chem. Commun., 2012, 48, 8637 RSC.
- W. Z. Yuan, Y. Q. Tan, Y. Y. Gong, P. Lu, J. W. Y. Lam, X. Y. Shen, C. F. Feng, H. H. Y. Sung, Y. W. Lu, I. D. Williams, J. Z. Sun, Y. M. Zhang and B. Z. Tang, Adv. Mater., 2013, 25, 2837 CrossRef CAS PubMed.
- R. Misra, T. Jadhav, B. Dhokale and S. M. Mobin, Chem. Commun., 2014, 50, 9076 RSC.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, J. Mater. Chem. C, 2015, 3, 9981 RSC.
- T. Jadhav, B. Dhokale, S. M. Mobin and R. Misra, RSC Adv., 2015, 5, 29878 RSC.
- Z. P. Yu, Y. Y. Duan, L. H. Cheng, Z. L. Han, Z. Zheng, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Mater. Chem., 2012, 22, 16927 RSC.
- G. Liu, M. D. Yang, L. K. Wang, J. Zheng, H. P. Zhou, J. Y. Wu and Y. P. Tian, J. Mater. Chem. C, 2014, 2, 2684 RSC.
- M. Chen, L. Z. Li, H. Nie, J. Q. Tong, L. L. Yan, B. Xu, J. Z. Sun, W. J. Tian, Z. J. Zhao, A. J. Qin and B. Z. Tang, Chem. Sci., 2015, 6, 1932 RSC.
- G. Yu, S. W. Yin, Y. Q. Liu, J. S. Chen, X. J. Xu, X. B. Sun, D. G. Ma, X. W. Zhan, Q. Peng, Z. G. Shuai, B. Z. Tang, D. B. Zhu, W. H. Fang and Y. Luo, J. Am. Chem. Soc., 2005, 127, 6335 CrossRef CAS PubMed.
- Z. Li, Y. Q. Dong, B. X. Mi, Y. H. Tang, M. Häussler, H. Tong, Y. P. Dong, J. W. Y. Lam, Y. Ren, H. H. Y. Sung, K. S. Wong, P. Gao, I. D. Williams, H. S. Kwok and B. Z. Tang, J. Phys. Chem. B, 2005, 109, 10061 CrossRef CAS PubMed.
- J. Mei, Y. N. Hong, J. W. Y. Lam, A. J. Qin, Y. H. Tang and B. Z. Tang, Adv. Mater., 2014, 26, 5429 CrossRef CAS PubMed.
- Z. M. Zhang, Y. Zhao, R. Zhang, L. F. Zhang, W. Q. Cheng and Z. H. Ni, Dyes Pigm., 2015, 118, 95 CrossRef CAS.
- Z. M. Zhang, F. F. Han, R. Zhang, N. Li and Z. H. Ni, Tetrahedron Lett., 2016, 57, 1917 CrossRef CAS.
- A. Kamkaew, S. H. Lim, H. B. Lee, L. V. Kiew, L. Y. Chung and K. Burgess, Chem. Soc. Rev., 2013, 42, 77 RSC.
- S. G. Awuah and Y. You, RSC Adv., 2012, 2, 11169 RSC.
- Y. C. Yang, Q. L. Guo, H. C. Chen, Z. K. Zhou, Z. J. Guo and Z. Shen, Chem. Commun., 2013, 49, 3940 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb and J. R. Cheeseman, Gaussian 09, revision A.02, Gaussian, Inc., Wallingford, CT, 2009 Search PubMed.
- Y. Y. Gong, Y. Q. Tan, J. Liu, P. Lu, C. F. Feng, W. Z. Yuan, Y. W. Lu, J. Z. Sun, G. F. He and Y. M. Zhang, Chem. Commun., 2013, 49, 4009 RSC.
- X. Zhou, H. Y. Li, Z. G. Chi, X. Zhang, J. Y. Zhang, B. J. Xu, Y. Zhang, S. W. Liu and J. R. Xu, New J. Chem., 2012, 36, 685 RSC.
- J. Mei, J. Wang, A. Qin, H. Zhao, W. Yuan, Z. Zhao, H. H. Y. Sung, C. Deng, S. Zhang, I. D. Williams, J. Z. Sun and B. Z. Tang, J. Mater. Chem., 2012, 22, 4290 RSC.
- B. J. Xu, Z. G. Chi, J. Y. Zhang, X. Q. Zhang, H. Y. Li, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, Chem.–Asian J., 2011, 6, 1470 CrossRef CAS PubMed.
- J. Kunzelman, M. Kinami, B. R. Crenshaw, J. D. Protasiewicz and C. Weder, Adv. Mater., 2008, 20, 119 CrossRef CAS.
- X. Q. Zhang, Z. G. Chi, J. Y. Zhang, H. Y. Li, X. F. Li, S. W. Liu, Y. Zhang and J. R. Xu, J. Phys. Chem. B, 2011, 115, 7606 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1478704 (2a), 1478706 (2b), 1478705 (2c), 1486489 (2d), crystal structures of compounds 2a–2d, AIE and mechanochromic properties of compounds 2b–2d, computational data and NMR spectra. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c6ra16514a |
| This journal is © The Royal Society of Chemistry 2016 |