
Theoretical study of the cis–trans isomerization mechanism of a pendant metal-bound azobenzene†
Ting-Ting Yin,
Zeng-Xia Zhao* and
Hong-Xing Zhang*
International Joint Research Laboratory of Nano-Micro Architecture Chemistry, Institute of Theoretical Chemistry, Jilin University, 130023 Changchun, China. E-mail: zhaozx@jlu.edu.cn; zhanghx@jlu.edu.cn; Tel: +86 18943121798
First published on 12th August 2016
Abstract
In this paper, we performed density-functional theory (DFT) at the B3LYP/6-311++G(d,p) & LANL2DZ quantum level to probe the thermal cis → trans isomerization of Re(CO)3–AB, a diimine ligand with an azo group substituent. We have searched for the transition states (TS) on the S0 and T1 potential energy surfaces and detected two inversion transition states TS-inv-1 and TS-inv-2 following a rotation-assisted inversion mechanism. In the T1 state, only the rotation pathway exists with the energy barrier 1.88 eV. We also investigated the photoisomerization pathway, the potential energy profiles of the vertical excitation for the excited states T1, S1, T2 and S2 were calculated, and we analyzed two possible isomerization routes through the inversion and rotation pathways. In addition, we probed the properties of the isomerization of the azo portion caused by excitation of the MLCT bands.
1. Introduction
Over the last few decades, the development of photo-responsive materials has become an intensive area of research, whose properties and eventual functionality are controlled by changes of the environment-light irradiation. These systems have been already implemented in an extensive range of modern materials and devices for daily applications like sunglass lenses, memory devices, photochromic inks and so forth.1–3 Transition metal compounds that show metal to ligand charge transfer transitions (MLCT), as seen in many d6 metal complexes such as rhenium, are perfect for transferring energy to an external group.4–9 Because of the importance and versatility of transition-metal complexes, the photochemical characters of rhenium tricarbonyl complexes with this type of ligands were extensively studied by Stufkens as early as in the 90's.10–12 And the focus of people's research is from how metal coordination could affect the photochromism of the organic switch located at the organic ligand shifted toward the modification of the properties and eventually the functionality of the metal complex upon isomerization of the photochromic fragment present in the ligands.6 Interest in this type of photoluminescent compounds remains strong as evidenced by a number of recent reviews.10,13,14 In principle, photo-responsive metal complexes can be acquired by incorporation of organic photochromic units in the structure of their ligands.1,15–17 Azobenzene (AB) is the photochromic fragment most frequently used for this purpose. AB does experience a reversible cis to trans photoisomerization which induces not only structural changes but also important electronic modifications in the ligands.18–26 The photoinduced isomerization of AB is a well-studied example of a light driven process.18,22,23,27 AB has been appended to a number of chromophores, including transition metal complexes, in an attempt to couple the cis → trans isomerization process to an external electronic transition.9,17,28–32Amazingly, in spite of the wide number of transition metal complexes of AB-containing Re(CO)3 have been investigated experimentally, however, concerning the thermal cis → trans isomerization there is not much known from the theoretical side. To the best of our knowledge, only the group of Hasheminasab and co-workers showed example of transition metal complex of AB-containing Re(CO)3 (Scheme 1).4 They designed and synthesized Re(CO)3–AB, shown UV-visible absorption bands resulted from both the AB unit and the Re(CO)3 diimine unit. Excitation of each compound led to isomerization of the azo-benzene bond, which they observed experimentally. But the data displayed here could not permit them to make comments on the nature of the AB isomerization (i.e., inversion vs. rotation). The fusion of the properties of transition metal complexes together with the photochromism has created a new generation of multifunctional metallic compounds able to react to an external stimuli, the light.33
 | ||
| Scheme 1 Photoisomerization of Re(CO)3–AB. | ||
Much theoretical work has been done to investigate the photochemistry of photo-triggered system, and has been proven (TD)DFT34,35 techniques are instrumental in interpreting photophysical behavior and spectra. DFT techniques with hybrid functionals are successfully used to visualize singlet and triplet CT states and reveal their characters by displaying differences of electron density distribution upon excitation and excited-state spin-density distributions.35–37 To demonstrate this photo-triggered process theoretically, density functional theory (DFT) and time-dependent (TD-DFT) calculations are performed to investigate the activity of a Re(CO)3 diimine complex with a pendant azo-benzene group at a molecular level. Here we wish to find out the most efficient isomerization routes in the S0 and T1 states according to a detailed study of their potential energy profiles of Re(CO)3–AB as well as discover how the organometallic group might influence the cis → trans isomerization of the pendant AB group. It may also act as a systemic supplement that provides valuable insights for future experimental studies of searching effective photo-responsive materials.
2. Computational details
2.1. Ground-state calculations
To understand the properties of the potential energy surfaces of Re(CO)3–AB, we conducted computational modeling of the trans and cis isomers of it. All the stable structures and the transition states for the thermal isomerization of Re(CO)3–AB in the S0 state were fully optimized unrestrainedly by density-functional theory (DFT) with the B3LYP38,39 (i.e., the long-range corrected version of the Becke's three-parameter hybrid exchange functional with the Lee–Yang–Parr correlation functional, using the Coulomb-attenuating method) functional. In another comparison test, several other functionals (CAM-B3LYP,40 M06,41 M06-2X41 and PBE0 (ref. 42)) were employed as supplements in order to avoid the possible shortcomings of DFT methods (Table S1 and Fig. S1†). All elements except rhenium were assigned the 6-311++G(d,p) basis set and the “double-ζ” quality basis set LANL2DZ with relativistic effective core potentials was employed for the rhenium atom based on the test of basis sets and computational costs consideration. These functional and basis sets have been justified in literature.43–45 To investigate the isomerization mechanism, the potential energy surfaces (PESs) were generated by scanning the N1N2C3 and the C5N1N2 angles from 90° to 280° at a 10.0° interval. The frequencies of all the constructions were computed at the same level to identify the nature of the stationary points and the transition states, and the zero-point energy (ZPE) corrections for each structure were obtained. Furthermore, the intrinsic reaction coordinate (IRC) theory was applied to identify the transition states connecting reactants and products.2.2. Excited-state calculations
Time dependent density-functional theory (TDDFT) with B3LYP/6-311++G(d,p) & LANL2DZ were used for the excited-state calculations as they were found to give authentic results.46,47 The excited-state potential energy surfaces were produced by counting single point vertical excitation energies for each of the points in the ground-state potential energy surfaces. All calculations were performed using Gaussian 09 suite of programs.48 Solvent interactions were simulated using a polarizable continuum model (PCM),49 with parameters taken from the water, acetonitrile, tetrahydrofuran, hexane and gas phase, for the sake of completeness. In order to compare with the experimental results, we chose THF at last.3. Results and discussion
3.1. Optimized ground-state geometry
The optimized structures of trans and cis isomers for Re(CO)3–AB in the S0 state have been illustrated in Fig. 1 and Table 1, and compared with the value of AB obtained in previous theoretical23,50–54 and experimental work (Table 1).55,56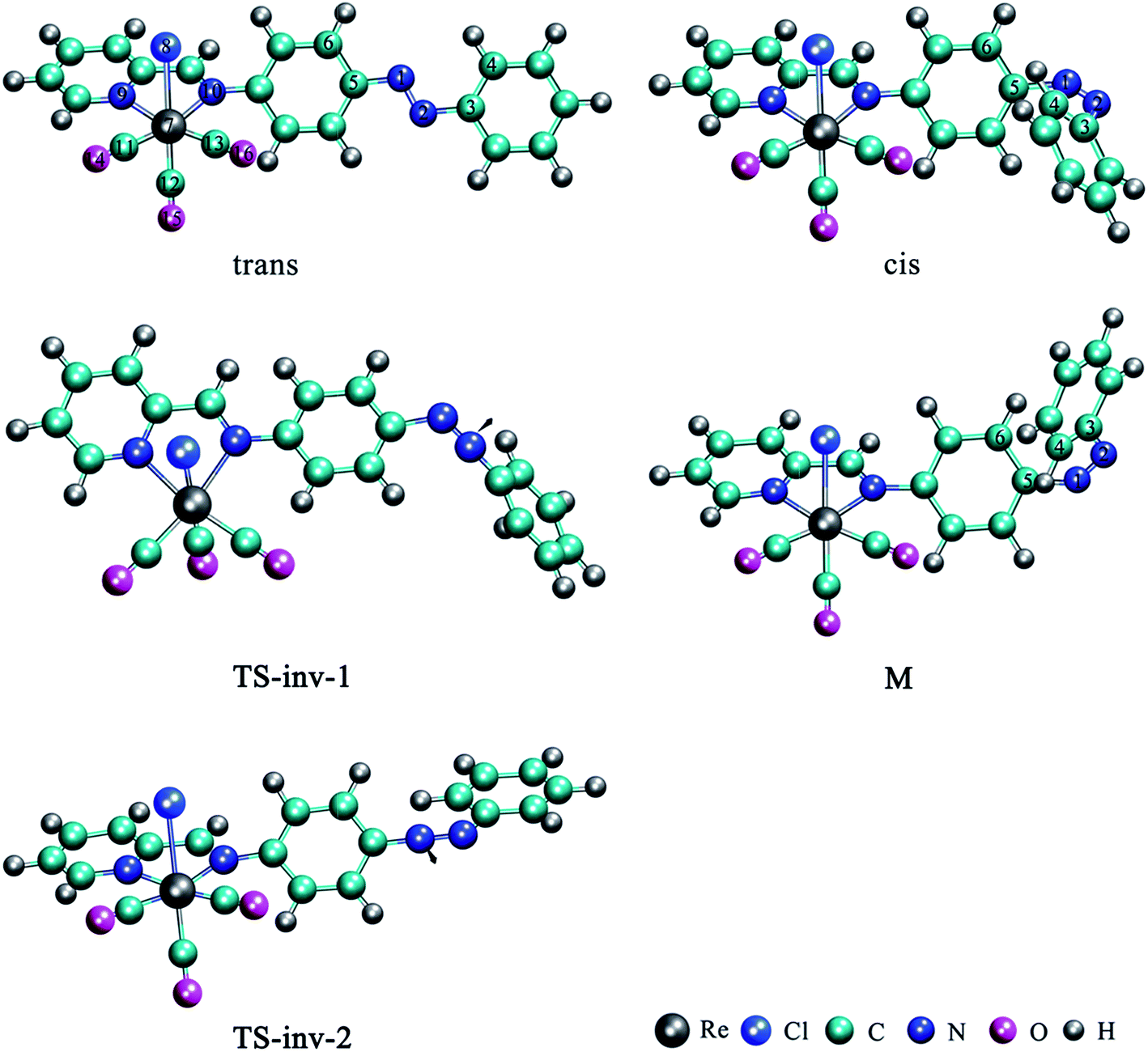 | ||
| Fig. 1 Optimized equilibrium geometries and structures of the two transition states (TS) of Re(CO)3–AB in the S0 state computed at B3LYP(PCM)/6-311++G(d,p) & LANL2DZ level. | ||
| Parameter | trans-S0 | cis-S0 | M | TS-inv-1 | TS-inv-2 | AB-trans/cisa | Exptb,c | trans-T1 | cis-T1 | TS-T1 |
|---|---|---|---|---|---|---|---|---|---|---|
| a Ref. 52.b Ref. 56.c Ref. 55. | ||||||||||
| R(C5N1) | 1.417 | 1.435 | 1.434 | 1.450 | 1.427 | 1.424/1.430 | 1.427/1.449 | 1.365 | 1.365 | 1.369 |
| R(N1N2) | 1.254 | 1.245 | 1.245 | 1.219 | 1.216 | 1.229/1.231 | 1.260/1.253 | 1.292 | 1.291 | 1.234 |
| R(N2C3) | 1.417 | 1.435 | 1.435 | 1.332 | 1.442 | 1.417/1.430 | 1.427/1.449 | 1.371 | 1.371 | 1.379 |
| R(Re7Cl8) | 2.545 | 2.546 | 2.547 | 2.545 | 2.552 | — | — | 2.546 | 2.546 | 2.551 |
| R(Re7N9) | 2.219 | 2.220 | 2.219 | 2.219 | 2.212 | — | — | 2.216 | 2.216 | 2.212 |
| R(Re7N10) | 2.225 | 2.225 | 2.224 | 2.222 | 2.244 | — | — | 2.235 | 2.234 | 2.246 |
| R(Re7C11) | 1.928 | 1.928 | 1.928 | 1.929 | 1.925 | — | — | 1.927 | 1.927 | 1.924 |
| R(Re7C12) | 1.915 | 1.914 | 1.915 | 1.915 | 1.912 | — | — | 1.914 | 1.914 | 1.913 |
| R(Re7C13) | 1.932 | 1.932 | 1.931 | 1.932 | 1.931 | — | — | 1.932 | 1.933 | 1.932 |
| R(C11O14) | 1.154 | 1.154 | 1.154 | 1.153 | 1.155 | — | — | 1.154 | 1.154 | 1.155 |
| R(C12O15) | 1.159 | 1.159 | 1.158 | 1.158 | 1.159 | — | — | 1.159 | 1.159 | 1.159 |
| R(C13O16) | 1.153 | 1.153 | 1.153 | 1.153 | 1.154 | — | — | 1.153 | 1.153 | 1.154 |
| A(C5N1N2) | 115.3 | 124.2 | 124.2 | 117.6 | 179.5 | 118.2/126.3 | 113.6/121.9 | 122.6 | 122.6 | 130.4 |
| A(N1N2C3) | 115.7 | 123.9 | 124.0 | 179.9 | 118.5 | 117.9/126.3 | 113.6/121.9 | 123.1 | 123.2 | 148.4 |
| D(C6C5N1N2) | −179.2 | 136.4 | 54.1 | −180.0 | 119.5 | 0.0/46.5 | 0.0/53.3 | −176.1 | 175.2 | 179.4 |
| D(C5N1N2C3) | −179.8 | −9.9 | 9.6 | 33.4 | −30.3 | 180.0/9.5 | 180.8/8.0 | −108.9 | 109.5 | 1.1 |
| D(N1N2C3C4) | 0.3 | −49.3 | 48.5 | −123.4 | 0.5 | 0.0/46.5 | 0.0/53.3 | 5.3 | 175.2 | −91.5 |
Analysis of the crystallographic data exhibits that Re(CO)3–AB crystallizes in the trans form with a N1N2 bond length of 1.254 Å that is almost identical to that of AB (average 1.25 Å), the C5N1N2C3 dihedral angle is −179.8°, while the N1N2C3C4 dihedral angle is 0.3°, so both the optimized metal diimine and AB groups are essentially planar, but these two units are non-coplanar. The non-planarity of cis-Re(CO)3–AB is instead substantial, with N1N2C3C4 = −49.3° and C5N1N2C3 = −9.9°. While the geometrical parameters of metal diimine group Re(CO)3 have almost no change in the two isomers. Table 2 displays the energy (E), the zero-point energy (ZPE) and the relative energy with respect to the trans-Re(CO)3–AB for all the species. The trans isomer is 0.59 eV more stable than the cis isomer, due to twisting of the benzene rings around the C–N bond to avoid steric hindrance, which is lower than that of AB reported in the literature.26,47,52,54,57,58 It indicates that binding metal diimine group to benzene rings for AB greatly increases the stability of the trans isomer, that is to say lowers the energy gap of trans and cis isomers. It is interesting to note that although there are small differences between gas-phase and solution-phase, especially in the values of the NNC and CNN angles (Table S2†), the geometries of cis and trans on the ground-state (S0) surface show an almost negligible dependency on the solvent, and the effect of the solvent on the cis-to-trans barrier height is rather small (Table S2†).
| E/a.u. | ZPE/a.u. | ΔEZPE/eV | |
|---|---|---|---|
| a ΔE is the relative energy with respect to the trans-S0.b Ref. 23.c Ref. 58.d Ref. 63.e Ref. 52. | |||
| trans-S0 | −1793.1738426 | 0.31010 | 0.00 |
| cis-S0 | −1793.1519706 | 0.30993 | 0.59 |
| M | −1793.1520029 | 0.31003 | 0.59 |
| TS-inv-1 | −1793.11056 | 0.30782 | 1.72 |
| TS-inv-2 | −1793.1176145 | 0.30811 | 1.48 |
| trans-T1 | −1793.130336 | 0.30813 | 1.13 |
| cis-T1 | −1793.1298957 | 0.30813 | 1.14 |
| TS-T1 | −1793.1020197 | 0.30722 | 1.88 |
| AB-trans/cisb | — | — | 0.71 |
| AB-trans/cisc | — | — | 0.70 |
| AB-trans/cisd | — | — | 0.80 |
| AB-trans/cise | — | — | 0.83 |
| AB-TSe | — | — | 2.28 |
3.2. Potential energy surface
Another point worthy of discussion is the potential energy surface (PES), numerous theoretical studies have been carried out to characterize the PES connecting the two possible isomers of AB (trans- and cis-AB). They expounded that the photoisomerization proceeds through torsion of the central CNNC fraction and planar inversion of the NNC angle either independently or simultaneously, or through hybridizations of torsion and inversion movement. For the rotation type mechanism, two dihedral angles CNNC and NNCC are the most significant variables. For the inversion type isomerization route, two bond angles NNC and CNN are the most important variables. For these points the varying angles were fixed, all the remaining parameters optimized, then the potential energy surfaces were carried out. We will discuss the two isomerization pathways separately in the following part by two-dimensional potential energy surfaces.The cis → trans isomerization can, in principle, proceed along different reaction pathways and through diverse transition states, enormous theoretical efforts have devoted to the study of the isomerization of ABs in gas and condensed phase, and also pointed out the solvent can affect the isomerization mechanism both in the ground and excited states.58–62
In accordance with previous studies, a transition state structure (TS-inv-1) (Fig. 1) was found to be responsible for inversion mechanism, being characterized by an almost linear N1N2C3 moiety (Table 1). Suitable identification of this construction as a true transition state was done, checking for the being of only one imaginary frequency in normal mode analysis. Based upon analysis, TS-inv-1 is assigned to the N2 stretching. Along this reaction path, the N1N2 distance decreases from the cis isomer to the TS-inv-1 and then increases in length as it approaches trans-S0. TS-inv-1 presents the strongest N1N2 double bond character along the trajectory. An in-plane inversion of the N1N2C3 angle is from 123.9° for cis-S0 to approximate 180° for TS-inv-1, while this angle is 115.7° for trans-S0. Inversion of the N1N2C3 angle does not cause obvious change of the C5N1N2 angle, only leads to a slight compression of N1N2 and N2C3 bonds and the elongation of C5N1 bond. As the cis-to-trans isomerization reaction proceeds through the inversion pathway, the N1N2C3 angle increases up to approximately 180° at the transition state. This is not to say that the rotation coordinate is frozen and it does not play any role. In fact, the C5N1N2C3 angle is an important parameter in defining the critical structures for the inversion mechanism, as it can be seen in Table 1. The torsional C5N1N2C3 angle is from −9.9° of cis-S0 to the value not far from 33.4° in the transition state, and −179.8° in the trans form. Therefore, this transition state is somewhat a mixture between inversion and rotational paths. And the hybridization of one nitrogen atom changes from sp2 to sp because the coupling between the two nitrogen lone-pairs weakens. There exists greater steric hindrance between the lone pairs on the central nitrogens and p orbitals of the phenyl ring close to the 180° N1N2C3 angle. From this perspective, the thermal cis–trans isomerization is not a pure inversion along NNC, but it follows a rotation-assisted inversion mechanism where the NNC angle must reach the value close to 180° but where the CNNC angle can take any value.
To have a better understanding of the potential energy profile, we calculated the reaction path from TS-inv-1 by scanning the N1N2C3 angle as shown in Fig. 2(a), which is started from the TS-inv-1 along the forward and reverse pathway, respectively. This transition state suggests that along the lowest energy path connecting the cis and trans isomers, these two structures are characterized, with respect to TS-inv-1, by a sort of motion of the perpendicular phenyl ring increases (cis-S0) or decreases (trans-S0) of the N1N2C3 angle, starting to define which will be the final isomers, upon surmounting a barrier of 1.13 eV on the potential energy surface, in AB this value is 2.05 eV.52 Manifesting that adding metal diimine group to the benzene rings of AB would lessen the cis–trans barrier height, making the thermal cis to trans isomerization easier than in AB. That is due to the MLCT effect minishes the energy demands for the isomerization reaction.
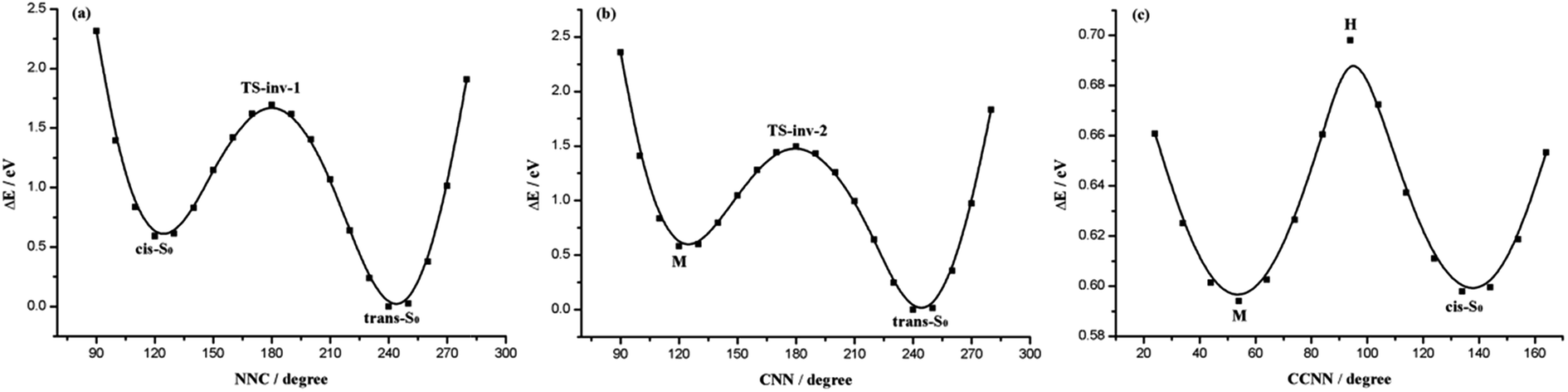 | ||
| Fig. 2 Potential energy profiles along the inversion of N1N2C3 angle (a), C5N1N2 angle (b) and rotation around C6C5N1N2 dihedral angle (c). | ||
Because the left and right side of this molecule is quite asymmetric, unlike AB and its derivatives have the same substitution of both sides, so we scan the C5N1N2 angle from cis isomer with the aim to determine the second inversion pathway see Fig. 2(b) and (c). An invertomer TS-inv-2 can be envisaged, with C5N1N2 angle constrained to be 179.1°, it is the transition state for trans → M belongs to the N1 stretching and the energy difference between the trans and M is 0.59 eV. Actually, this transition state is found at slightly lower energy, 1.48 eV, compared with TS-inv-1. It is visualized from the PES (Fig. 2(c)) that there is an energy barrier during the rotation around the C6C5N1N2 axis from M to cis-S0, requiring energy of about 0.11 eV (Table 2) for M to cis. The optimized geometries of M show little variation in the N1N2 bond length and the most bond parameters, but show overt variations in the C6C5N1N2 and C5N1N2C3 dihedral angles.
We have also tried to optimize the rotational transition state, looking for a hypothetical transition state geometry, such a transition state might influence the thermal isomerization mechanism. The conclusion is negative, although we tried a number of different initial structures for the TS-rot to obtain the saddle point for the rotational pathway in THF, we could not get a stationary point along that path, neither in water, in acetonitrile, in hexane and in gas phase. But we can find the inversion transition states (only tested TS-inv-1) in all the above condition except acetonitrile, geometrical parameters and energies are depicted in Table S3.†
Based on the above discussion, we can present an intuitive point of view about the global thermal isomerization pathways of Re(CO)3–AB. The whole process of cis → trans isomerization for Re(CO)3–AB can be depicted as follows: the transition state of the overall reaction is connected to two equilibrium structures cis-S0 and trans-S0, respectively, by the inversion of the N1N2C3 angle. However, another transition state can link to intermediate M and trans-S0 by the inversion of the C5N1N2 angle. Through an energy barrier around 0.11 eV reaching cis isomer from M by the rotation of the C6C5N1N2 dihedral angle. This isomerization mechanism can thus be accounted in terms of successive rotation, inversion, and rotation processes. In the isomerization process, the TS-inv-1(S0) and TS-inv-2(S0) structures can only be changed as the N1N2C3 and C5N1N2 inversion increase or decrease, because the Re(CO)3–AB molecule needs to remain its symmetry. At the same time, its change is also restricted by the metal complex (Re(CO)3 moiety) in the symmetrical position. Therefore, the thermal inversion isomerization process involves the rotation of the phenyl ring.
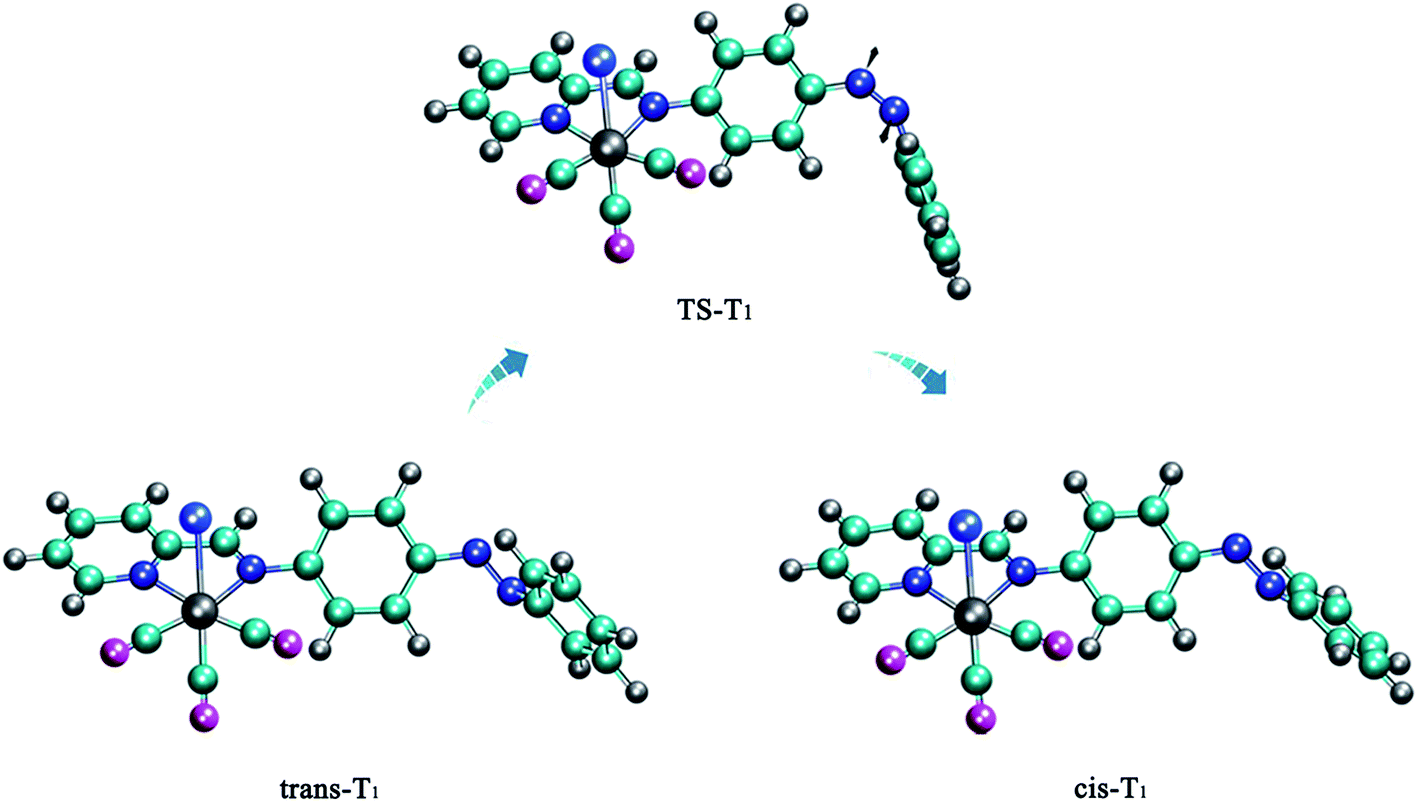 | ||
| Fig. 3 Optimized structures of all the stationary points of the T1 state. | ||
Next we studied the potential energy profile of T1 state (Fig. 4). The geometries and energies of the two minimum points on the potential energy profile are consistent with trans-T1 and cis-T1. And we show the construction of the transition state TS-T1 in Fig. 3, which is characterized by C5N1N2C3 = 1.1° (Table 1). We find the two phenyl rings of TS-T1 are almost perpendicular, and the N1N2 double bond character still remains throughout the isomerization process. The barrier height for this isomerization is 1.88 eV (Table 2), which is determined by subtracting from the energy of trans-T1. It exhibits an obvious preference for the rotation route. Accordingly, the Re(CO)3–AB molecule can isomerize via the rotation manner at trans or cis point when it is excited to the T1 state.
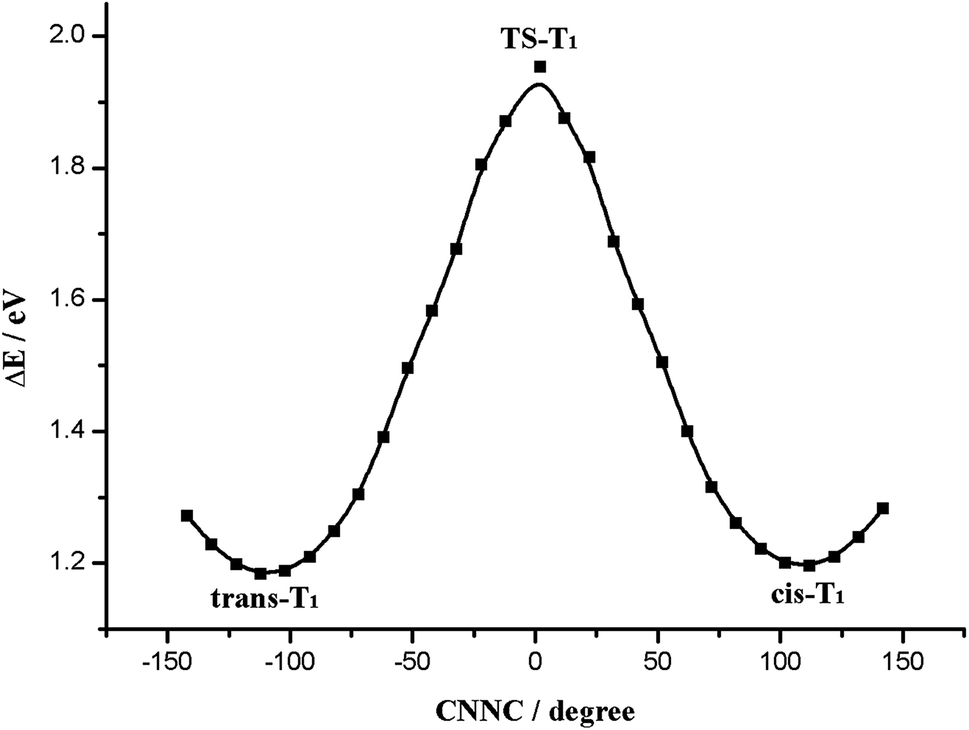 | ||
| Fig. 4 Potential energy profile of the T1 state along the C5N1N2C3 rotation coordinate. | ||
3.3. Isomerization pathways of excited states
Electronic energies (E) and oscillator strengths (f) of the compound was computed by means of time dependent DFT calculations at the B3LYP(PCM)/6-311++G(d,p) & LANL2DZ level of theory. The data associated with the main transitions computed for this compound are presented in Table 3, and the shape of the involved frontier molecular orbitals are depicted in Fig. 5.| Electron state | Key excitation | Character | E (eV)/λ (nm) | f | |
|---|---|---|---|---|---|
| a Ref. 58.b Ref. 52.c Ref. 26. | |||||
| trans | S0 → S1 | H → L (96%) | dπ(Re) → L(π*) | 2.29 (542) | 0.0004 |
| S0 → S2 | H−3 → L (54%) | Ln → L(π*) | 2.48 (500) | 0.0009 | |
| H−3 → L+1 (42%) | Ln → L(π*) | ||||
| S0 → T1 | H−3 → L (33%) | Ln → L(π*) | 1.79 (692) | 0.0000 | |
| H−3 → L+1 (59%) | Ln → L(π*) | ||||
| S0 → T2 | H−4 → L (22%) | dπ(Re) → L(π*) | 2.05 (606) | 0.0000 | |
| H−2 → L (44%) | Lπ → L(π*) | ||||
| H−2 → L+1 (20%) | Lπ → L(π*) | ||||
| H → L (19%) | dπ(Re) → L(π*) | ||||
| cis | S0 → S1 | H−1 → L (90%) | dπ(Re) → L(π*) | 2.33 (531) | 0.0004 |
| S0 → S2 | H → L (59%) | dπ(Re) → L(π*) | 2.43 (510) | 0.1407 | |
| H → L+1 (26%) | dπ(Re) → L(π*) | ||||
| S0 → T1 | H → L (18%) | dπ(Re) → L(π*) | 1.62 (765) | 0.0000 | |
| H → L+1 (70%) | dπ(Re) → L(π*) | ||||
| S0 → T2 | H−1 → L (58%) | dπ(Re) → L(π*) | 2.18 (569) | 0.0000 | |
| H → L (14%) | dπ(Re) → L(π*) | ||||
| AB-trans/cisa | S0 → S1 | — | n → π* | 3.24 (383)/3.36 (369) | — |
| AB-trans/cisb | S0 → S1 | — | n → π* | 3.08 (403)/3.77 (329) | — |
| AB-trans/cisc | S0 → S1 | — | n → π* | 2.53 (490)/2.72 (456) | — |
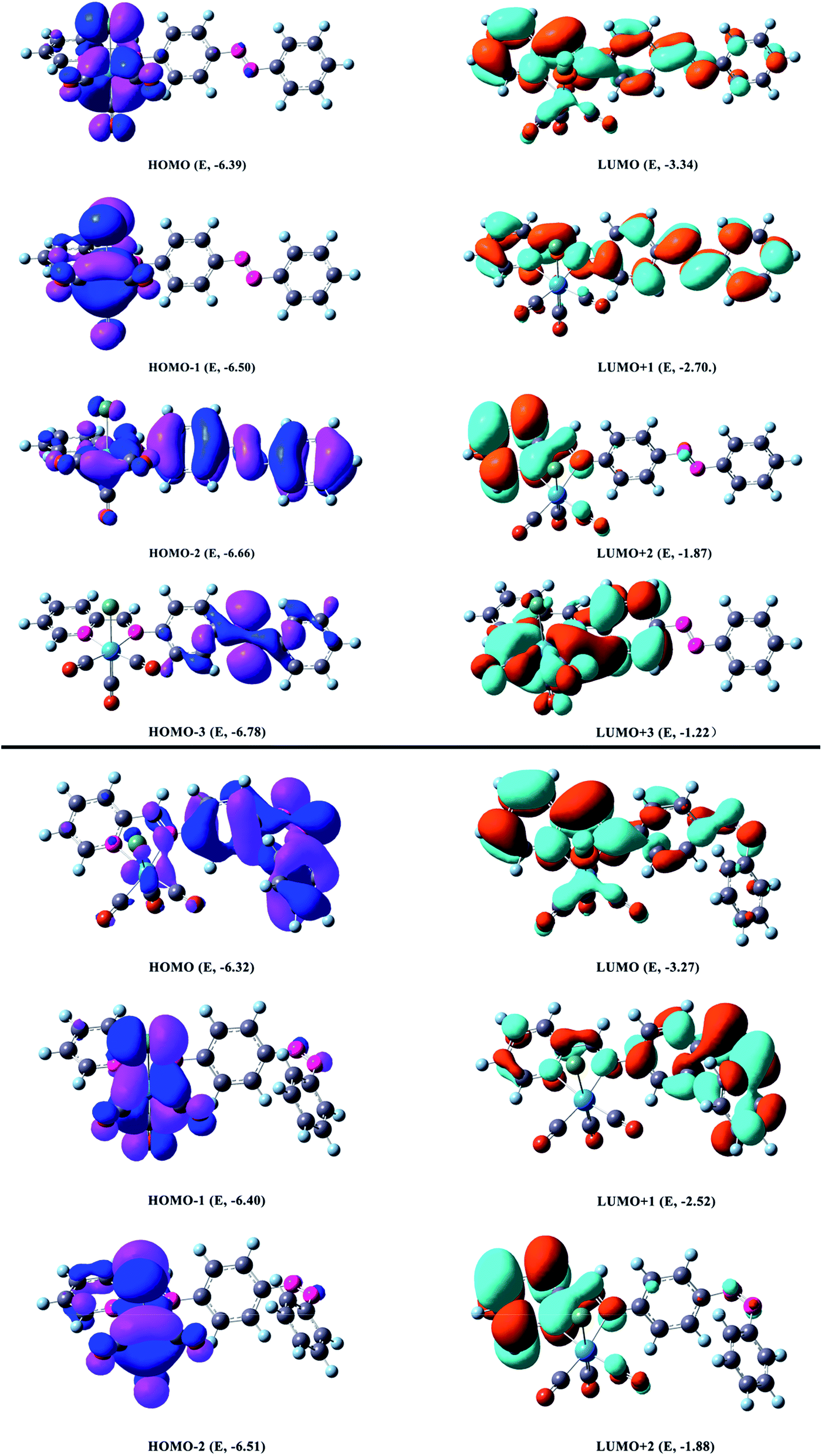 | ||
| Fig. 5 Most relevant MOs associated with the vertical excitations and its energy in eV shown in Table 4. Occupied and unoccupied orbitals are represented by purple & blue or yellow & green surfaces respectively (top: trans form, bottom: cis form). | ||
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N– moiety (34%) and π*(L) (66%).
N– moiety (34%) and π*(L) (66%).
| Orbital | Energy (eV) | % of composition | Main bond type | |||
|---|---|---|---|---|---|---|
| Re | CO | Ligand | ||||
| NN |
Aromatic system | |||||
| L+1 | −2.584 | 3 | 2 | 35 | 60 | π*(L) |
| L | −3.259 | 5 | 4 | 8 | 83 | π*(L) |
| H | −6.339 | 53 | 19 | 0 | 27 | dπ(Re) + π*(CO) + π(L) |
| H−1 | −6.445 | 52 | 17 | 0 | 31 | dπ(Re) + π*(CO) + π(L) |
| H−2 | −6.584 | 16 | 6 | 12 | 66 | dπ(Re) + π*(L) |
| H−3 | −6.677 | 0 | 0 | 34 | 66 | n(L) + π*(L) |
| H−4 | −6.890 | 64 | 20 | 2 | 13 | dπ(Re) + π(CO) + π*(L) |
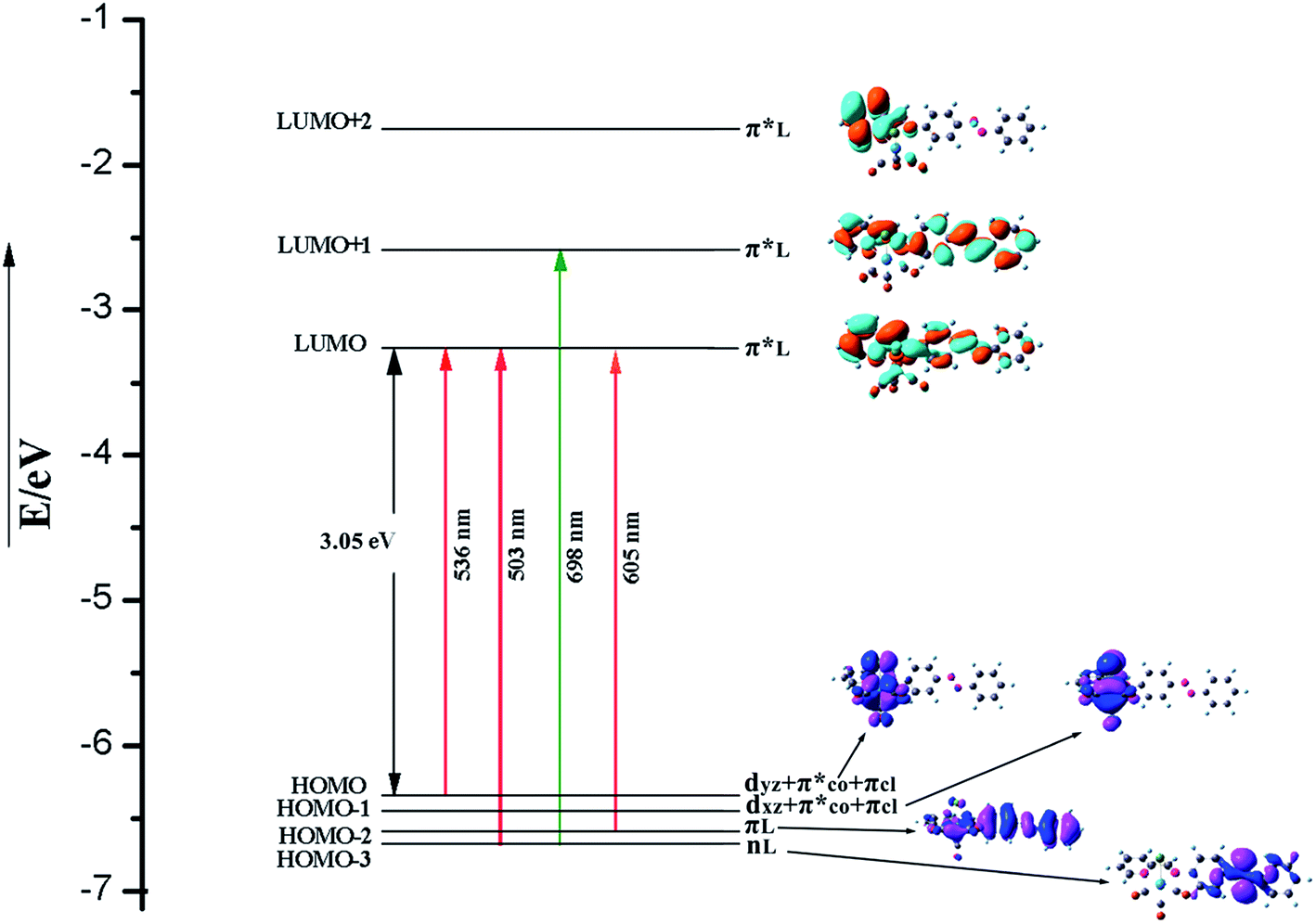 | ||
| Fig. 6 Energy level diagram of MOs involved in singlet–singlet and singlet–triplet vertical excitations of trans form. | ||
The orbital energies nearest the initial H–L gap are displayed in Fig. 5 and 6 (trans form), and one can see that the H and L energies are strongly affected by the Re(CO)3 group. The substitution pattern has also an important effect on the computed H–L gaps. Our results show that the electron withdrawing group Re(CO)3 substituted compound present a gap of about 3.05 eV lower than the pure AB counterpart 3.86 eV.64
Encouraged by the results from above, we shifted our focus to the isomerization pathways of excited states, we calculated the ground state single point vertical excitation energy to obtain the T1, S1, T2 and S2 states potential energy profiles along the inversion and rotating ways.
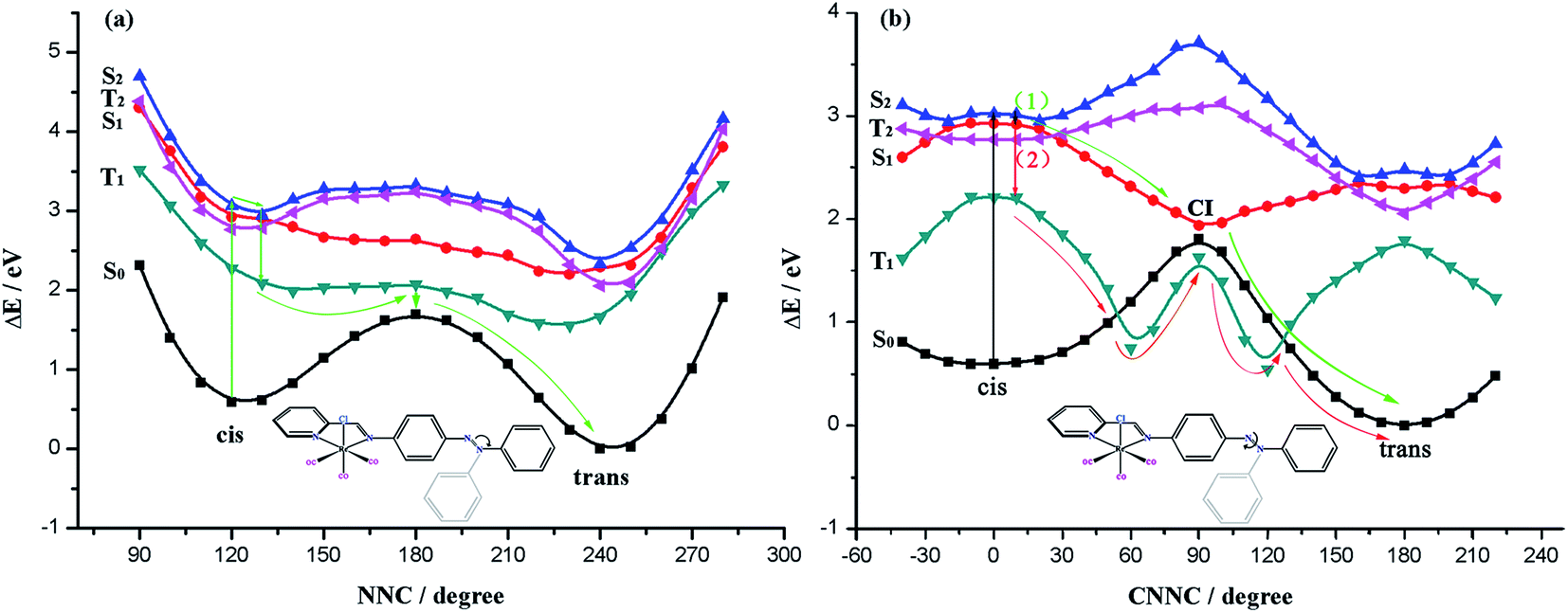 | ||
| Fig. 7 Potential energy profiles in the excited states for Re(CO)3–AB along the N1N2C3 inversion coordinate (a) and the C5N1N2C3 rotation coordinate (b). | ||
Along the inversion route, the possible isomerization pathway can be depicted as follows: cis-isomer would absorb light (hν) excited to the S2 state from S0 state and reached to the minimum point of S2 state. Because there is a high energy barrier of S2, so it can only be relaxed to a lower energy state for isomerization reaction to occur. Although the lifetime of the T2 state is long, the transition to the T2 state is forbidden, the possibility of occurrence of such intersystem crossing is less likely, thus relaxes to the S1 minimum fast. The potential energy surfaces of S1 and T1 states are very close, it may achieve this transition through spin–orbit coupling effect. Along the T1 state potential energy surface, near the maximum value close to the S0 state, through intersystem crossing to the potential energy profile of S0 state and gets to the trans isomer. The isomerization pathways are indicated by the arrow in Fig. 7(a). Along the isomerization pathway of inversion coordinates, rapid distribution of energy would occur due to repeated vibrational relaxation, when this process is completed, little energy is left to the coordinates associated with isomerization, leading to a low isomerization efficiency, and therefore the chance of this reaction is vanishingly small.
The CNN potential energy surface is depicted in Fig. S2,† by compared the potential energy curves with NNC, we find there is no significant difference, so we don't discuss here.
Intriguingly, we found that the maximum of T1 state potential energy profile is not on top of the energy barrier of S0 state, but below the maximum of S0 state energy profile, this is because in the vicinity of the maximum, it forms a biradical system with the breakage of N1N2 bond, therefore, T1 state is more stable at this time. Such a feature might influence the thermal isomerization mechanism. Due to the fact that S0 state's potential energy surface would visit T1 state at the dihedral angle C5N1N2C3 spinning area, isomerization process can be realized through the S0–T1–S0, making the thermodynamic isomerization go through a recession of T1 ← S0 intersystem crossing. This isomerization process has a low energy barrier of 1.88 eV, but modification of the spin multiplicity is needed.
The calculations suggest that along the isomerization way of coordinate-rotation begins with absorbing light energy of cis isomer excited to S2 state from S0 state, because there is a high energy barrier of S2 state, therefore it reaches the nadir of S1 state potential energy profile after releasing its energy through fast vibration relaxation. With this, there are two pathways: (1) along the S1 state potential energy profile, it can transit to the maximum of S0 state potential energy surface without radiation by way of the CI of the S1 and S0 states potential energy surfaces, subsequently gains isomerization product cis isomer along the S0 state potential energy profile; (2) from S1 state to T1 state by intersystem crossing, then reaches isomerization product along the potential energy profile of T1 state by the intersection of S0 and T1 states (Fig. 7(b)).
4. Concluding remarks
In this work, we have theoretically studied the influence brought by the additional metal complex on AB isomerization and analyzed the thermal cis → trans isomerization mechanism of Re(CO)3–AB.Calculations reveal that the energy transfer process of metal complex occurs due to partial diazo antibonding character of the LUMO orbital, allowing the MLCT band to induce isomerization. We have found that binding metal diimine group to benzene rings for AB drastically lowers the energy barrier of the cis → trans isomerization, making it easier to isomerize. It is concluded that the thermal isomerization of Re(CO)3–AB mainly follows the NNC rotation-assisted inversion mechanism in the S0 state. We also searched the potential energy profiles of the vertical excitation for the excited states (T1, S1, T2 and S2) along the inversion and rotation pathways. By detailed analysis, we presented a hypothesis that can be accomplished in the following steps: (1) the isomerization can easily occur through the S0/S1 conical intersection (CI) and descend to the S0 state; (2) a relaxation to the T1 state from the S1 state may occur and the reaction could take place via the S0–T1–S0 path.
These complicating factors must be addressed in designing more efficient switching in organo rhenium complexes. We are continuing our work on searching effective photo chemical materials.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (Grant No. 21173096) and the State Key Development Program for Basic Research of China (Grant No. 2013CB834801).Notes and references
- J. Pérez-Miqueo, A. Telleria, M. Muñoz-Olasagasti, A. Altube, E. García-Lecina, A. de Cózar and Z. Freixa, Dalton Trans., 2015, 44, 2075–2091 RSC.
- H. Bouas-Laurent and H. Dürr, Pure Appl. Chem., 2001, 73, 639–665 CrossRef CAS.
- J. Żmija and M. Małachowski, Issues, 2010, 1, 2 Search PubMed.
- A. Hasheminasab, L. Wang, M. Dawadi, J. Bass, R. Herrick, J. Rack and C. Ziegler, Dalton Trans., 2015, 44, 15400–15403 RSC.
- M. Busby, P. Matousek, M. Towrie and A. Vlček, Inorg. Chim. Acta, 2007, 360, 885–896 CrossRef CAS.
- C. C. Ko and V. W. W. Yam, J. Mater. Chem., 2010, 20, 2063–2070 RSC.
- T. Yutaka, I. Mori, M. Kurihara, J. Mizutani, K. Kubo, S. Furusho, K. Matsumura, N. Tamai and H. Nishihara, Inorg. Chem., 2001, 40, 4986–4995 CrossRef CAS PubMed.
- A. Sakamoto, A. Hirooka, K. Namiki, M. Kurihara, M. Murata, M. Sugimoto and H. Nishihara, Inorg. Chem., 2005, 44, 7547–7558 CrossRef CAS PubMed.
- M. E. Moustafa, M. S. McCready and R. J. Puddephatt, Organometallics, 2012, 31, 6262–6269 CrossRef CAS.
- M. S. Jana, A. K. Pramanik, S. Kundu, D. Sarkar and T. K. Mondal, Inorg. Chim. Acta, 2013, 399, 138–145 CrossRef CAS.
- B. D. Rossenaar, D. J. Stufkens and A. Vlček, Inorg. Chem., 1996, 35, 2902–2909 CrossRef CAS.
- B. D. Rossenaar, E. Lindsay, D. J. Stufkens and A. Vlček, Inorg. Chim. Acta, 1996, 250, 5–14 CrossRef CAS.
- I. R. Farrell and A. N. Vlček, Coord. Chem. Rev., 2000, 208, 87–101 CrossRef CAS.
- K. K. W. Lo, W. K. Hui, C. K. Chung, K. H. K. Tsang, T. K. M. Lee, C. K. Li, J. S. Y. Lau and D. C. M. Ng, Coord. Chem. Rev., 2006, 250, 1724–1736 CrossRef CAS.
- O. S. Wenger, Chem. Soc. Rev., 2012, 41, 3772–3779 RSC.
- H. Nishihara, Coord. Chem. Rev., 2005, 249, 1468–1475 CrossRef CAS.
- M. Kurihara and H. Nishihara, Coord. Chem. Rev., 2002, 226, 125–135 CrossRef CAS.
- H. D. Bandara and S. C. Burdette, Chem. Soc. Rev., 2012, 41, 1809–1825 RSC.
- P. Cattaneo and M. Persico, Phys. Chem. Chem. Phys., 1999, 1, 4739–4743 RSC.
- L. Duarte, R. Fausto and I. Reva, Phys. Chem. Chem. Phys., 2014, 16, 16919–16930 RSC.
- F. Zapata, M. Á. Fernández-González, D. Rivero, Á. Álvarez, M. Marazzi and L. M. Frutos, J. Chem. Theory Comput., 2014, 10, 312–323 CrossRef CAS PubMed.
- T. Schultz, J. Quenneville, B. Levine, A. Toniolo, T. J. Martínez, S. Lochbrunner, M. Schmitt, J. P. Shaffer, M. Z. Zgierski and A. Stolow, J. Am. Chem. Soc., 2003, 125, 8098–8099 CrossRef CAS PubMed.
- A. Cembran, F. Bernardi, M. Garavelli, L. Gagliardi and G. Orlandi, J. Am. Chem. Soc., 2004, 126, 3234–3243 CrossRef CAS PubMed.
- A. W. Adamson, A. Vogler, H. Kunkely and R. Wachter, J. Am. Chem. Soc., 1978, 100, 1298–1300 CrossRef CAS.
- C. W. Chang, Y. C. Lu, T. T. Wang and E. W. G. Diau, J. Am. Chem. Soc., 2004, 126, 10109–10118 CrossRef CAS PubMed.
- I. Conti, M. Garavelli and G. Orlandi, J. Am. Chem. Soc., 2008, 130, 5216–5230 CrossRef CAS PubMed.
- G. Tiberio, L. Muccioli, R. Berardi and C. Zannoni, ChemPhysChem, 2010, 11, 1018–1028 CrossRef CAS PubMed.
- T. Yutaka, I. Mori, M. Kurihara, J. Mizutani, N. Tamai, T. Kawai, M. Irie and H. Nishihara, Inorg. Chem., 2002, 41, 7143–7150 CrossRef CAS PubMed.
- K. Yamaguchi, S. Kume, K. Namiki, M. Murata, N. Tamai and H. Nishihara, Inorg. Chem., 2005, 44, 9056–9067 CrossRef CAS PubMed.
- T. Yutaka, I. Mori, M. Kurihara, N. Tamai and H. Nishihara, Inorg. Chem., 2003, 42, 6306–6313 CrossRef CAS PubMed.
- C. J. Lin, Y. H. Liu, S. M. Peng and J. S. Yang, J. Phys. Chem. B, 2012, 116, 8222–8232 CrossRef CAS PubMed.
- R. Sakamoto, S. Kume, M. Sugimoto and H. Nishihara, Chem.–Eur. J., 2009, 15, 1429–1439 CrossRef CAS PubMed.
- A. Telleria, J. Pérez-Miqueo, A. Altube, E. García-Lecina, A. de Cózar and Z. Freixa, Organometallics, 2015, 34, 5513–5529 CrossRef CAS.
- M. Turki, C. Daniel, S. Záliš, A. Vlček Jr, J. van Slageren and D. J. Stufkens, J. Am. Chem. Soc., 2001, 123, 11431–11440 CrossRef CAS PubMed.
- A. Cannizzo, A. M. Blanco-Rodríguez, A. Nahhas, J. Sebera, S. Záliš, A. Vlček Jr and M. Chergui, J. Am. Chem. Soc., 2008, 130, 8967–8974 CrossRef CAS PubMed.
- A. M. Blanco-Rodríguez, A. Gabrielsson, M. Motevalli, P. Matousek, M. Towrie, J. Sebera, S. Záliš and A. Vlček Jr, J. Phys. Chem. A, 2005, 109, 5016–5025 CrossRef PubMed.
- A. Gabrielsson, M. Busby, P. Matousek, M. Towrie, E. Hevia, L. Cuesta, J. Perez, S. Záliš and A. Vlček Jr, Inorg. Chem., 2006, 45, 9789–9797 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- P. Stephens, F. Devlin, C. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623–11627 CrossRef CAS.
- T. Yanai, D. P. Tew and N. C. Handy, Chem. Phys. Lett., 2004, 393, 51–57 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- C. Adamo and V. Barone, J. Chem. Phys., 1999, 110, 6158–6170 CrossRef CAS.
- K. Potgieter, T. Gerber, R. Betz, L. Rhyman and P. Ramasami, Polyhedron, 2013, 59, 91–100 CrossRef CAS.
- V. Baranovskii and D. Maltsev, Comput. Theor. Chem., 2014, 1043, 71–78 CrossRef CAS.
- R. Sarkar and K. K. Rajak, J. Org. Chem., 2015, 779, 1–13 CrossRef CAS.
- K. Ichimura, Photochromic materials and photoresists, Photochromism: Molecules and Systems, ed. H. Dürr and H. Bouas-Laurent, Elsevier, Amsterdam, 1990, pp. 903–918 Search PubMed.
- C. R. Crecca and A. E. Roitberg, J. Phys. Chem. A, 2006, 110, 8188–8203 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, N. J. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09 Revision D.01, Gaussian, Inc, Wallingford, CT, 2009 Search PubMed.
- V. Barone, M. Cossi and J. Tomasi, J. Chem. Phys., 1997, 107, 3210–3221 CrossRef CAS.
- H. Fliegl, A. Köhn, C. Hättig and R. Ahlrichs, J. Am. Chem. Soc., 2003, 125, 9821–9827 CrossRef CAS PubMed.
- M. Quick, A. L. Dobryakov, M. Gerecke, C. Richter, F. Berndt, I. N. Ioffe, A. A. Granovsky, R. Mahrwald, N. P. Ernsting and S. A. Kovalenko, J. Phys. Chem. B, 2014, 118, 8756–8771 CrossRef CAS PubMed.
- L. Yu, C. Xu and C. Zhu, Phys. Chem. Chem. Phys., 2015, 17, 17646–17660 RSC.
- M. Pederzoli, J. Pittner, M. Barbatti and H. Lischka, J. Phys. Chem. A, 2011, 115, 11136–11143 CrossRef CAS PubMed.
- R. L. Klug and R. Burcl, J. Phys. Chem. A, 2010, 114, 6401–6407 CrossRef CAS PubMed.
- T. Tsuji, H. Takashima, H. Takeuchi, T. Egawa and S. Konaka, J. Phys. Chem. A, 2001, 105, 9347–9353 CrossRef CAS.
- D. L. Beveridge and H. Jaffe, J. Am. Chem. Soc., 1966, 88, 1948–1953 CrossRef CAS.
- N. Kurita, S. Tanaka and S. Itoh, J. Phys. Chem. A, 2000, 104, 8114–8120 CrossRef CAS.
- M. Pederzoli, J. Pittner, M. Barbatti and H. Lischka, J. Phys. Chem. A, 2011, 115, 11136–11143 CrossRef CAS PubMed.
- J. C. Corchado, M. L. Sanchez, I. F. Galvan, M. E. Martín, A. M. Losa, R. B. Morgado and M. A. Aguilar, J. Phys. Chem. B, 2014, 118, 12518–12530 CrossRef CAS PubMed.
- T. Cusati, G. Granucci, E. M. Nunez, F. Martini, M. Persico and S. Vazquez, J. Phys. Chem. A, 2012, 116, 98–110 CrossRef CAS PubMed.
- J. Dokic, M. Gothe, J. Wirth, M. V. Peters, J. Schwarz, S. Hecht and P. Saalfrank, J. Phys. Chem. A, 2009, 113, 6763–6773 CrossRef CAS PubMed.
- L. M. Tiago, S. I. Beigi and S. G. Louie, J. Chem. Phys., 2005, 122, 094311 CrossRef PubMed.
- N. Kurita, S. Tanaka and S. Itoh, J. Phys. Chem. A, 2000, 104, 8114–8120 CrossRef CAS.
- Y. J. Lee, S. I. Yang, D. S. Kang and S. W. Joo, Chem. Phys., 2009, 361, 176–179 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra10880f |
| This journal is © The Royal Society of Chemistry 2016 |