
Low-voltage carbon films deposition by electro-exfoliation of graphite into graphene oxide†

Abstract
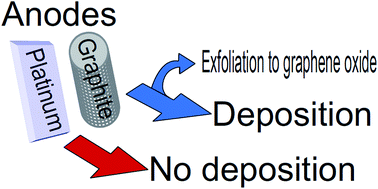
Low-voltage carbon films deposition by electro-exfoliation of graphite into graphene oxide is reported. By simply employing two different anodes, Pt or graphite, it is demonstrated that the carbon film deposition takes place by the graphite electro-exfoliation and not by the carbonic molecules from the electrolyte as has been frequently reported. By means of Raman spectroscopy, scanning electron microscopy (SEM), cyclic voltammetry (CV), X-ray diffractometry (XRD), atomic force microscopy (AFM), and dynamic light scattering (DLS), graphene oxide is successfully characterized. In addition, this electro-exfoliation process may be an alternative green route for the production of graphene oxide.
 Please wait while we load your content...
Please wait while we load your content...

