
Chemical modification of starch with epoxy resin to enhance the interfacial adhesion of epoxy-based glass fiber composites
Abstract
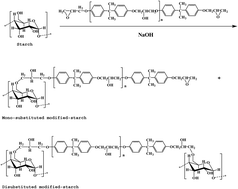
In order to fabricate epoxy-based glass fiber composites with superior mechanical and thermal properties, starch was chemically modified by E-51 epoxy resin, as a sizing for glass fibers. The hydrophilicity of starch was enhanced after modification as the surface tension decreased from 64.98 to 47.99 mN m−1, and the contact angle between the starch suspension and slide glass decreased from 50.26° to 33.53°. Besides, the interfacial adhesion between glass fiber and epoxy resin was obviously improved with the help of the modified starch, which can be clearly observed from SEM images. Consequently, significant increases of tensile strength from about 35 MPa to over 57 MPa, bending strength from about 65 MPa to over 87 MPa, and impact strength from about 8 KJ m−2 to over 14 KJ m−2 were obtained. Moreover, with the improvement of interfacial adhesion between the modified-starch sized glass fiber and epoxy resin, the thermo-stability of the composite was also improved as demonstrated by DSC. This study suggested a simple but effective chemical modification technique using a modifier to enhance interfacial adhesion in fabricating epoxy-based glass fiber composites with superior properties.
 Please wait while we load your content...
Please wait while we load your content...

