
Sugarcane molasses as a pseudocapacitive material for supercapacitors†

Abstract
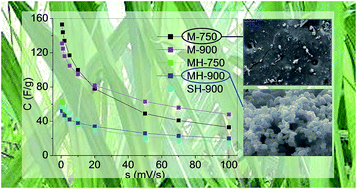
Oxygen-rich carbons were obtained from sugarcane molasses by two methods: direct carbonisation on one hand, and hydrothermal carbonisation and subsequent pyrolysis on the other hand. As no activation treatments were applied, the porous texture was poorly developed and mainly composed of ultramicropores with restricted access to electrolyte ions. Despite this, the directly carbonised molasses exhibited specific capacitances up to 153 F g−1 at 0.5 mV s−1 in 1 M H2SO4 electrolyte when tested as electrodes in a three-electrode system. Given the low specific surface areas of the carbons, the capacitance values were mainly assigned to their pseudocapacitance contributions. The latter were more adequately estimated by considering the NLDFT surface area (SNLDFT) than the BET area (ABET) due to the narrow porosity of the materials. Maximum values of pseudocapacitance contribution of 35.2% were attained for the carbon with a SNLDFT of 178 m2 g−1, which were explained by the high concentrations of surface oxygen groups, such as quinones and carbonyls.
 Please wait while we load your content...
Please wait while we load your content...

