
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of PEGylation and non-ionic surfactants on the physical stability of the therapeutic protein filgrastim (G-CSF)
Patrick K.
Chang
,
Clive A.
Prestidge
,
Timothy J.
Barnes
and
Kristen E.
Bremmell
*
School of Pharmacy and Medical Sciences, Sansom Research Institute, University of South Australia, Adelaide, South Australia 5000, Australia. E-mail: Kristen.bremmell@unisa.edu.au; Tel: +61 8 83022311
First published on 15th August 2016
Abstract
Improvement in the in vitro and in vivo stability of biotherapeutic proteins has been approached via a number of strategies, including protein PEGylation or formulation with non-ionic surfactants. Here we report on interaction and stability studies for the biotherapeutic protein filgrastim (granulocyte stimulating factor (G-CSF)) and its PEGylated analogue (PEG-GCSF), with polysorbate 20, using isothermal calorimetry, circular dichroism, surface tension and dynamic light scattering measurements. PEGylation of G-CSF did not alter temperature-induced conformational changes detected with circular dichroism, however did increase the amphiphilic nature of G-CSF, lowering the surface tension to a greater extent. G-CSF and PEG-GCSF both aggregated at temperatures below that of denaturation. G-CSF had an inverse relationship between concentration and the temperature at which aggregation was initiated, with aggregates continually increasing in size to greater than 2 μm. Importantly, PEG-GCSF was shown to have improved resistance to heat-induced aggregation; the presence of PEG attached to the protein minimised the aggregate size to below 120 nm. Interaction between polysorbate 20 and the proteins was weak and determined to result from a hydrophobic mechanism. A two-site binding model was found to best describe the interaction of polysorbate 20 with G-CSF, irrespective of PEGylation. Presence of polysorbate 20 did not minimise the thermal-induced instability for G-CSF or PEG-GCSF. These findings provide new insight into the mechanism of therapeutic protein stabilization using PEG and non-ionic surfactants.
1. Introduction
Therapeutic proteins offer distinct advantages over other small drug molecules, such as having a specific mechanism of action, high potency, and bioactivity even at low therapeutic concentrations. As a result, these molecules have become one of the fastest growing classes of therapeutic molecules on the market.1,2 The native protein 3D conformation is a product of its specific amino acid sequence and the presence of secondary structures such as α-helices and β-sheets. Due to the inherent structure–activity relationship of proteins, successfully maintaining the specific conformation is critically important during their production, transportation and storage.One approach to address formulation stability of the native protein conformation and potential for adsorption or aggregation has been to formulate bio-therapeutics with non-ionic surfactants. Their role in stabilising therapeutic protein formulations has not been well established, due in some part to the protein dependent nature of the mechanism. A number of studies have investigated the effect of non-ionic surfactants, in particular polysorbates, on therapeutic protein stability and also on the interaction between polysorbates and proteins. Both the influence on stability and the binding of polysorbates to proteins has been demonstrated to be dependent on the type of protein. Non-ionic surfactants have been shown to interact with proteins by acting as a chemical chaperone, binding to the protein and preventing protein aggregation and adsorption to surfaces, as encountered during transportation and storage of proteins where agitation and exposed interfaces may induce aggregation.3,4 Surfactant molecules may bind to exposed hydrophobic patches present on the surface of the protein molecule5–7 and have been shown to favour refolding of protein over self-induced aggregation by binding transiently with partially unfolded proteins.3,8 Above the critical micelle concentration (CMC), the presence of surfactant micelles may aid in protection of proteins by encapsulating the protein within the micelle, which prevents self-induced aggregation or interaction between protein molecules with hydrophobic interfaces.1 Preferential adsorption of surfactant at air–solution and solution–solid interfaces, preventing protein adsorption, denaturation and aggregation at these interfaces has also been proposed.9
Protein conjugation with polyethylene glycol (PEG), a water soluble, biocompatible polymer is now a well-established method to enhance the in vivo circulation lifetime and reduce the frequency of dosing required for protein therapeutics, with a number of PEGylated proteins on the market.10,11 Biological activity and circulation time of PEGylated proteins have been reported extensively in literature, with fewer studies reporting on formulation and stability. Conjugation of the hydrated PEG chain results in an increased molecular volume, contributing to the longer circulation time and consequently impacting on some of the biophysical properties of the protein. Most studies have reported the secondary and tertiary structure to be independent of PEGylation.12,13 In terms of stability in solution, the conformation and aggregation of proteins has been demonstrated to depend on both the PEG conjugation strategy and the protein. Protein stability in solution has been demonstrated to increase for various PEGylated proteins; for example, PEG-GCSF exhibited a reduced rate and extent of aggregate formation compared to its non-PEGylated form, while retaining a similar aggregation pathway,14 and the thermal stability human growth hormone was increased dependent on the conjugation method.15 PEGylated lysozyme was also found to have lower propensity toward aggregate formation compared to native lysozyme.16
Granulocyte Colony Stimulating Factor (G-CSF) is a cytokine that regulates the proliferation and differentiation of neutrophilic granulocytes, possessing functional activities on granulocytes and monocyte-macrophages.17,18 Recombinant G-CSF has been marketed for clinical use in enhancing hematopoietic recovery after cancer chemotherapy and bone marrow transplantation.19,20 Expressed from Escherichia coli (E. coli), G-CSF is a non-glycosylated protein consisting of 175 amino acid residues with a molecular weight of 18.8 kDa. The sequence of this isolated protein is structurally identical to the sequence of G-CSF isolated from human blood, except for the presence of methionine at the N-terminal residue, necessary for both expression in E. coli, as well as for chemical conjugation of polyethylene-glycol (PEG).21 PEG-GCSF exhibits comparable clinical outcomes to G-CSF, with a reduced dosing cycle as a result of its long circulation half-life.11 While PEGylated protein therapeutics are formulated with non-ionic surfactants, similarly to non-PEGylated analogues, few studies have considered the impact of non-ionic surfactants on the physical stability of PEGylated proteins. Treuheit et al.22 reported increased thermal degradation of PEG-GCSF with increased concentration of polysorbate 20 over a 59 week study, however for agitation induced aggregation, increased polysorbate 20 concentrations decreased the aggregation.
In this study, we investigated both the impact of PEGylation on biophysical stability aspects of G-CSF, and the interaction of the non-ionic surfactant, polysorbate 20, with G-CSF and PEG-GCSF. Circular dichroism (CD) was used to provide information on conformational changes with respect to PEGylation and in the presence of polysorbate 20. Surface tension measurements were used to probe the influence of PEGylation on G-CSF adsorption to the air–water interface, providing knowledge on the amphiphilic nature of the proteins. Isothermal titration calorimetry (ITC) was used to investigate the nature of binding between surfactant and protein, and dynamic light scattering was used to monitor protein aggregation as a function of temperature. This provides important insight into protein formulation and stability using PEGylation and non-ionic surfactants.
2. Materials and methods
2.1. Materials
Pharmaceutical grade human recombinant Granulocyte Colony Stimulating Factor (G-CSF), PEGylated Granulocyte Colony Stimulating Factor (PEG-GCSF) and pure polysorbate 20 used in this study were provided by Hospira. Inc, Adelaide. The G-CSF component is a small globular protein (∼18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 800 Da) composed of 4 α-helices and 2 disulfide bonds. The PEG was a 20 kDa mPEG chain covalently attached to the N terminus of G-CSF. The proteins were obtained as stock solutions in 10 mM sodium acetate buffer at pH 4, with G-CSF and PEG-GCSF concentration of 3.5 mg ml−1 and 10 mg ml−1, respectively. Acetic acid, Glacial (AR grade) was obtained from Ajax Chemicals and sodium acetate salt purchased from Sigma (Australia). Milli-Q water was used throughout.
800 Da) composed of 4 α-helices and 2 disulfide bonds. The PEG was a 20 kDa mPEG chain covalently attached to the N terminus of G-CSF. The proteins were obtained as stock solutions in 10 mM sodium acetate buffer at pH 4, with G-CSF and PEG-GCSF concentration of 3.5 mg ml−1 and 10 mg ml−1, respectively. Acetic acid, Glacial (AR grade) was obtained from Ajax Chemicals and sodium acetate salt purchased from Sigma (Australia). Milli-Q water was used throughout.
2.2. Methods
The binding stoichiometry measurements were controlled at a constant temperature of 25.0 ± 0.02 °C. The 1.4 ml sample cell was filled with protein solution diluted to the desired concentration (0.5 mg ml−1) with 10 mM pH 4.0 sodium acetate buffer prepared previously and the injection syringe (nominal volume 250 μl) was filled with polysorbate 20 solution (1.5% w/v). All titration experiments consisted of 25 injections of 10.0 μl in 30 seconds with a spacing time of 300 seconds to allow appropriate time for temperature to equilibrate back to baseline. The initial delay for all experiments was set to 60 seconds with reference power and filter set to 10.0 μcal s−1 (corresponding to 42 μJ s−1) and 2 seconds, respectively. Stirring speed was set to 300 RPM (average revolution per minute) to ensure continuous mixing efficiency. The solution in the sample cell was continuously stirred while titrating 10 μL of surfactant to prevent adsorbed layers of protein or surfactant being formed in the ITC experiment.24 A similar protocol was followed for the temperature study at 25, 40 and 50 °C, with a 0.1 mg ml−1 protein solution used.
Titration curves were analysed using Origin® software provided by Microcal, LLC. The data were corrected for the surfactant's heat of dilution by performing dilution of surfactant to buffer, to obtain only heat signatures that are associated with the surfactant–protein interaction. Data from the first injections were also discarded before fitting of data, due to abnormal results that may arise from a volumetric error caused by backlash in the motorized screw used to drive the syringe plunger.25
3. Results and discussion
3.1. Bio-physical properties of G-CSF and the influence of PEGylation
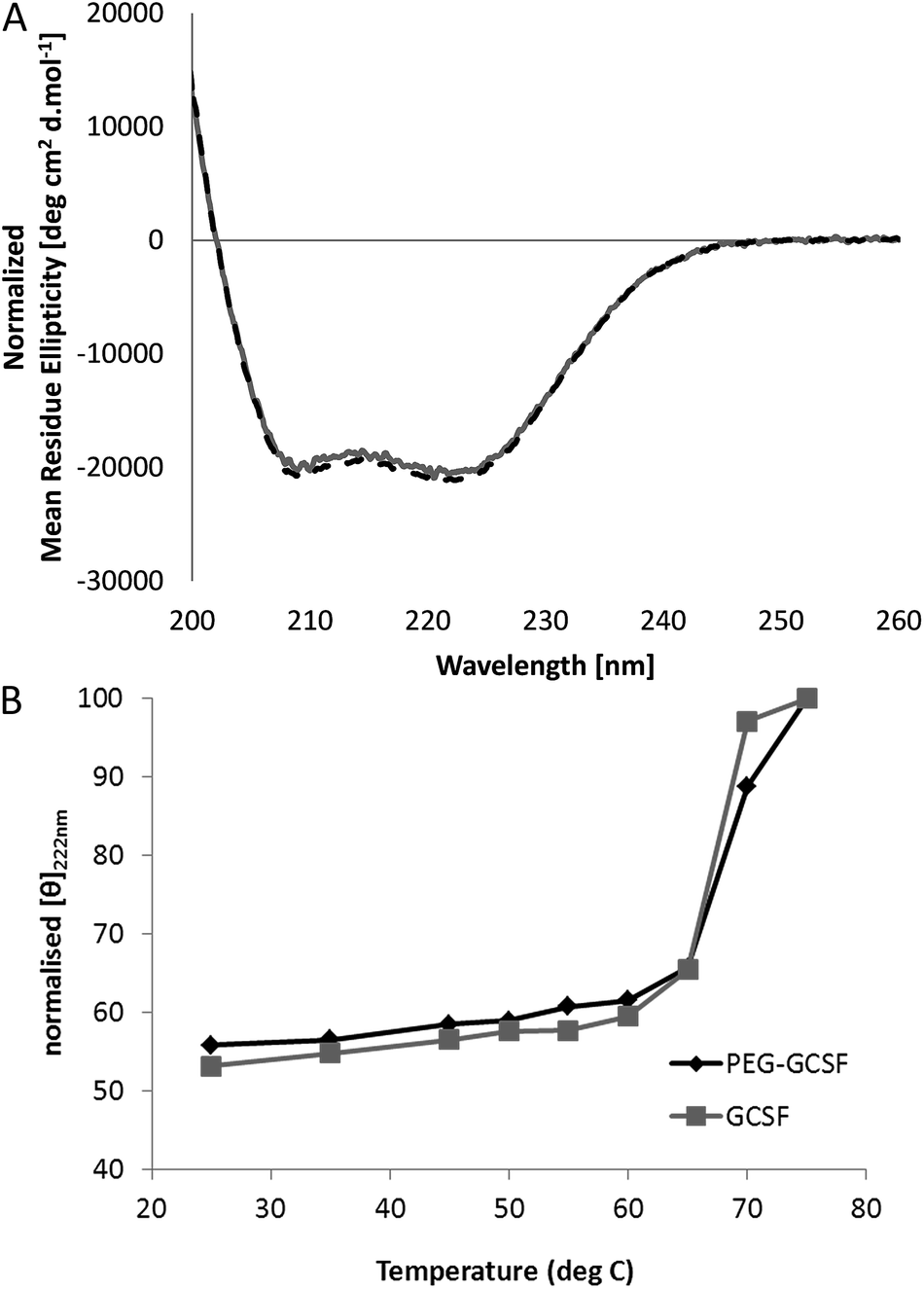 | ||
| Fig. 1 (A) Influence of PEGylation on the CD spectrum of G-CSF at 25 °C (G-CSF: grey line; PEG-GCSF: black dashed line), and (B) monitoring the intensity of the circular dichroism spectra at 222 nm indicates the melting process for G-CSF and PEG-GCSF. All measured in pH 4 acetate buffer. | ||
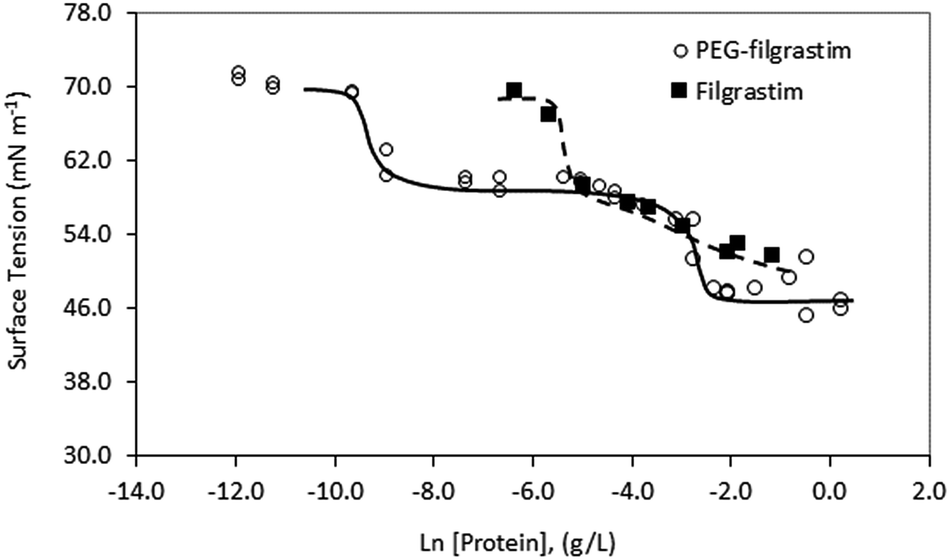 | ||
| Fig. 2 Surface tension of G-CSF and PEG-GCSF solutions as a function of concentration, in pH 4 acetate buffer. | ||
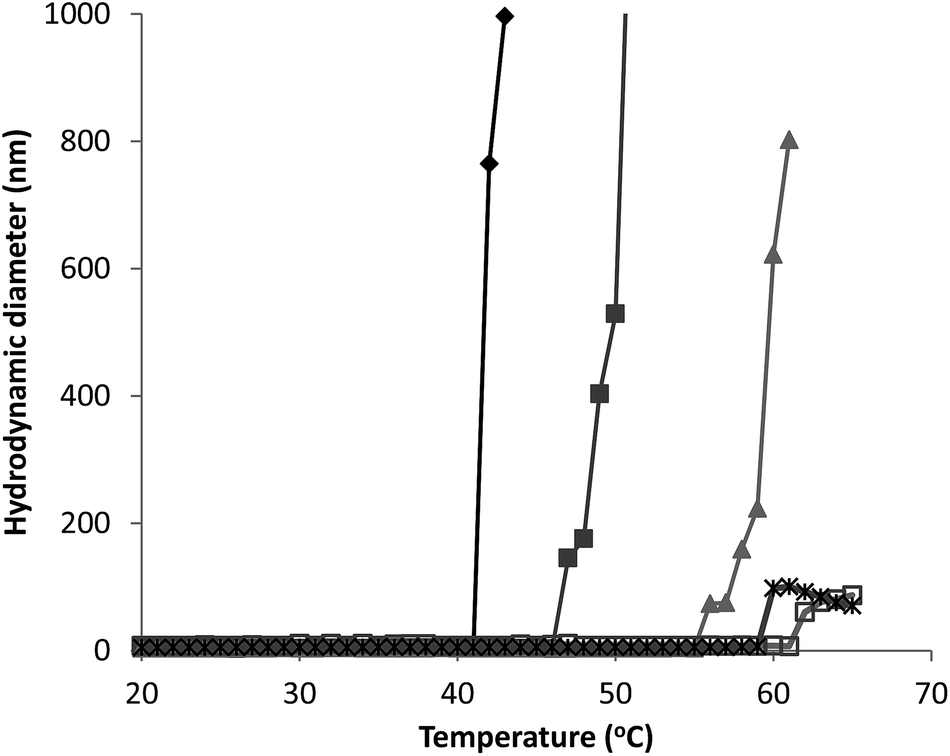 | ||
Fig. 3 Hydrodynamic diameter as a function of temperature for G-CSF (0.5 ( ), 1.5 ( ), 1.5 ( ) and 3.5 ( ) and 3.5 ( ) mg mL−1) and PEG-GCSF (1 ( ) mg mL−1) and PEG-GCSF (1 ( ) and 5 ( ) and 5 ( ) mg mL−1) in 10 mM acetate buffer at pH 4. ) mg mL−1) in 10 mM acetate buffer at pH 4. | ||
Irrespective of PEGylation, both proteins demonstrated aggregation at temperatures below that of the denaturation temperature measured with CD. The melting temperature determined from CD represents a global structural transformation within proteins and does not provide information on smaller conformational changes that may be taking place at temperatures less than the melting point. Other techniques have also suggested the 3-dimensional structure of G-CSF to undergo conformational changes at temperatures lower than required for denaturation. In a study by Rajan et al.,14 increasing conformational transition from alpha-helix to β-sheet structure was observed for both G-CSF and PEG-GCSF with increasing temperature using second-derivative FTIR. In addition, a recent time-of-flight secondary ion mass spectrometry (ToF-SIMS) study that investigated conformational aspects of adsorbed G-CSF as a function of temperature, detailed continuous changes to the 3-dimensional structure as temperature increased from 25 to 55 °C.31 Amino acid residues of tyrosine, methionine and phenylalanine, all of which contain hydrophobic functional groups, were found to contribute to the observed structural change and provided evidence for exposure of hydrophobic moieties of G-CSF at temperatures as low as 35 °C. All of these observations are consistent with exposure of hydrophobic amino acid sequences of G-CSF at temperatures below that required for macroscopic denaturation, which would allow for greater association between protein molecules leading to aggregation, as detected here using dynamic light scattering.
Significantly, PEGylation of G-CSF modified the extent of aggregation, greatly hindering aggregate growth. While G-CSF demonstrated continually growing aggregates to sizes greater than 2 μm, PEG-GCSF aggregates remained at sizes less than 120 nm. This confirms the work by Rajan et al.,14 where conjugation of PEG to G-CSF was found to reduce the size of aggregates, and in a study of lysozyme, PEGylation was found to reduce the formation of lysozyme particles by half.16 Therefore, while PEGylation did not alter conformational changes with increased temperature, it did infer stability against thermal induced aggregation, suggesting protection of exposed hydrophobic regions by the hydrated PEG chain. This aspect is significant; while PEGylation could not protect the protein from denaturation with increasing temperature, it has been shown to limit aggregation to only a few protein molecules, which is beneficial from a therapeutic formulation aspect, in limiting the growth size of protein aggregates.
3.2. Surfactant interactions and G-CSF stability
Despite evidence for increased stability against aggregation, commercial PEGylated therapeutic proteins continue to be formulated using non-ionic surfactants, similarly to their non-PEGylated analogues. With few studies examining interactions and stability of non-ionic surfactant/PEGylated protein formulations, it is therefore interesting to investigate the effect of PEGylation on the role and interactions of non-ionic surfactants in protein formulations. From surface tension measurements discussed earlier (Fig. 2), both G-CSF and PEG-GCSF are amphiphilic, with the presence of hydrophobic regions providing potential sites for both self-association and interaction with hydrocarbon chains of surfactant molecules. In this section, the influence of non-ionic surfactant, polysorbate 20, on the structural and physical stability of G-CSF and PEG-GCSF in solution will be explored.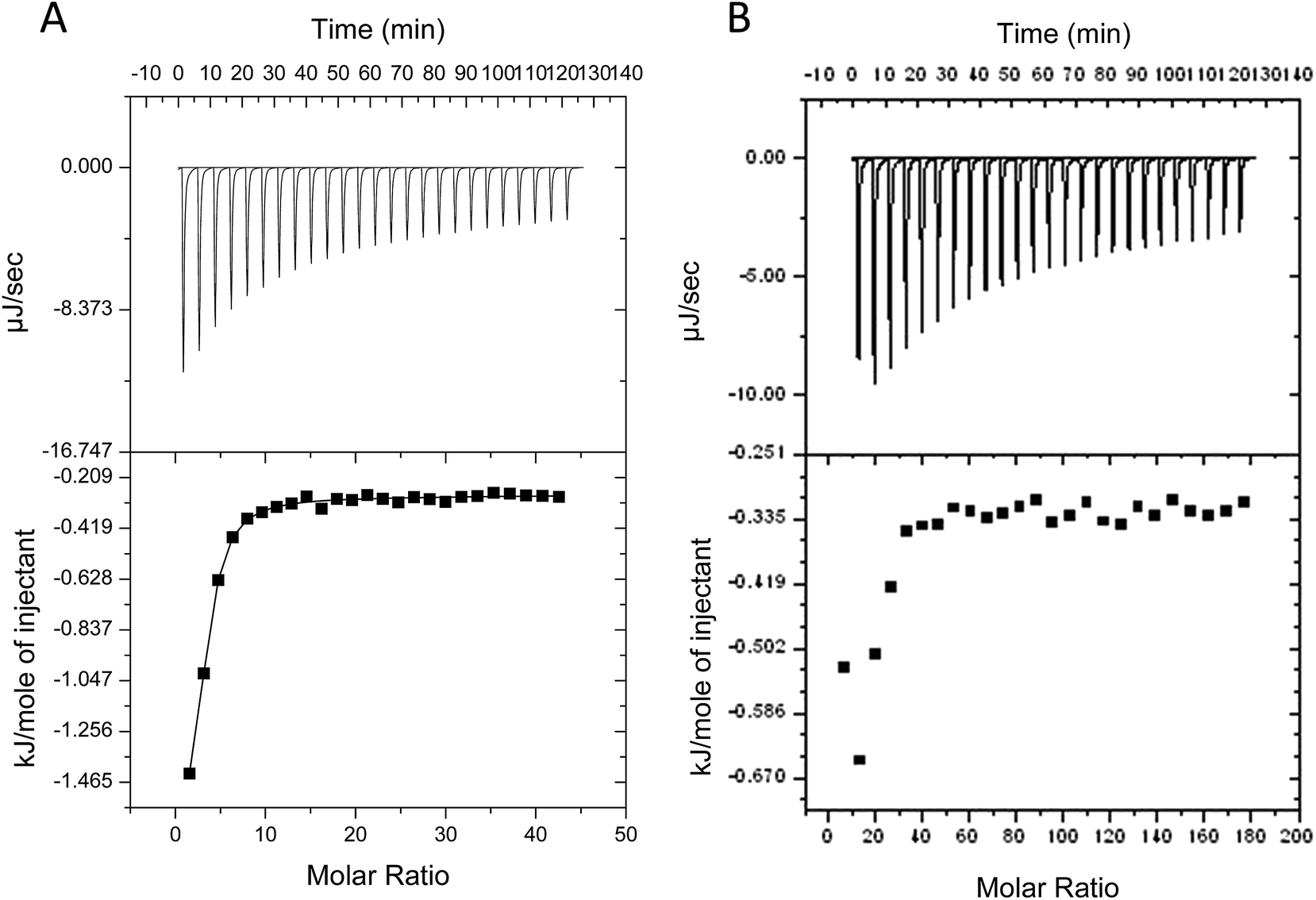 | ||
| Fig. 4 Experimental power flow signals (top), and associated binding isotherms (bottom) for the calorimetric titrations of polysorbate 20 to 0.5 mg ml−1 G-CSF (A) and 0.5 mg ml−1 PEG-GCSF (B). Thermograms were corrected for surfactant heat of dilution associated with each polysorbate 20 injection. Solutions were prepared and diluted in 10 mM sodium acetate buffer. | ||
All titration experiments lead to a low change heat and this was exothermic in nature. Multiple titration experiments were performed prior to obtaining the final results as shown in Fig. 4, with different concentration of both polysorbate 20 and proteins in 10 and 200 mM sodium acetate buffer. All resulted in similar thermograms that are characterized by low binding heats that are not representative of typical binding curves due to the absence of a baseline at low polysorbate to protein ratios. This suggests that only a fraction of polysorbate 20 monomers bind to G-CSF and that the binding is weak. However, calculation of the binding enthalpy (ΔH) and binding affinity (K) from the titration data of polysorbate 20 to both G-CSF and PEG-GCSF was attempted, with a two site kinetic model consistently providing the best fit (Table 1). The data were fitted with the assumption that the binding of polysorbate 20 to both proteins occur at multiple sites, with two sets of binding affinities indicating binding occurred with two different states (surfactant monomers and aggregates). The observation that polysorbate 20 interacts with G-CSF in two different mechanisms is supported by research measuring the kinetics of surface tension depression by mixed polysorbate/G-CSF solutions,32 where it was concluded that G-CSF associated with both surfactant aggregates and individual molecules.
| Protein | Binding constant Ka (M−1) | Enthalpy ΔH (kJ mol−1) | ||
|---|---|---|---|---|
| 1st | 2nd | 1st | 2nd | |
| G-CSF | 4.4 × 105 | 3.0 | −4.6 | −0.3 |
| PEG-GCSF | 1.2 × 104 | 23.8 | 12.8 | −66.4 |
Polysorbate 20 has also shown to exhibit similar binding behaviour producing two different binding affinities with other proteins, such as to albutropin.4 Albutropin is a novel form of human Growth Hormone (hGH), genetically fused with human serum albumin (HSA) with 6 alpha-helical structures in each domain.33 Previous studies have also demonstrated similar surfactant binding behaviour such as the interaction of sodium dodecyl sulfate (SDS) to bovine serum albumin (BSA), showing two classes of SDS binding sites.34 Salmaso et al.,35 also concluded the binding of G-CSF with PEG-cholane (an amphiphilic compound) was multi modal in nature, with a two site binding model providing the best fit to the ITC data.
Interaction of polysorbate 20 with PEG-GCSF was similarly studied with ITC (Fig. 4B). As a result of protein concentration limitations, data were difficult to obtain, and similarly to G-CSF lacked a baseline at low polysorbate 20 to PEG-GCSF ratios. However, the form of the isotherm was best fitted with a two site model, similar to that of G-CSF and suggest the interaction behaviour between polysorbate 20 and G-CSF is similar, irrespective of PEGylation. A recent study investigating PEGylated lysozyme also concluded that PEGylation had little impact on the nature of interaction of lysozyme with excipients such as sucrose and guanidine hydrochloride.36 The isotherm for PEG-GCSF suggests the interaction to be even weaker than that observed with G-CSF, with low binding affinities determined (Table 1). Intuitively, a structural interference to any interaction of polysorbate 20 with the PEG-GCSF molecule would be inferred by the presence of the 20 kDa PEG chain attached to the protein.
Binding isotherms for the interaction of polysorbate 20 to 0.1 mg ml−1 solutions of G-CSF and PEG-GCSF were measured by ITC between temperatures of 25 and 55 degrees (Fig. 5), below the onset of thermal denaturation measured by CD. With increased temperature, it can be seen for both systems that there was a decrease in the reaction enthalpy from 25 °C to 40 °C (Fig. 5). The decrease in enthalpy observed was similar to that observed by Hoffmann et al.,37 and was associated with an entropic effect reducing the protein–surfactant interaction. However at 55 °C, both proteins showed an increase in negative reaction enthalpy, indicating an increase in surfactant–protein binding. This may have occurred as a result of expansion of the proteins native conformation exposing hydrophobic regions of the protein as discussed earlier and in Kempson et al.,31 for increased binding of the hydrophobic surfactant chains. This provides further evidence for polysorbate 20 interaction with G-CSF via a hydrophobic mechanism.
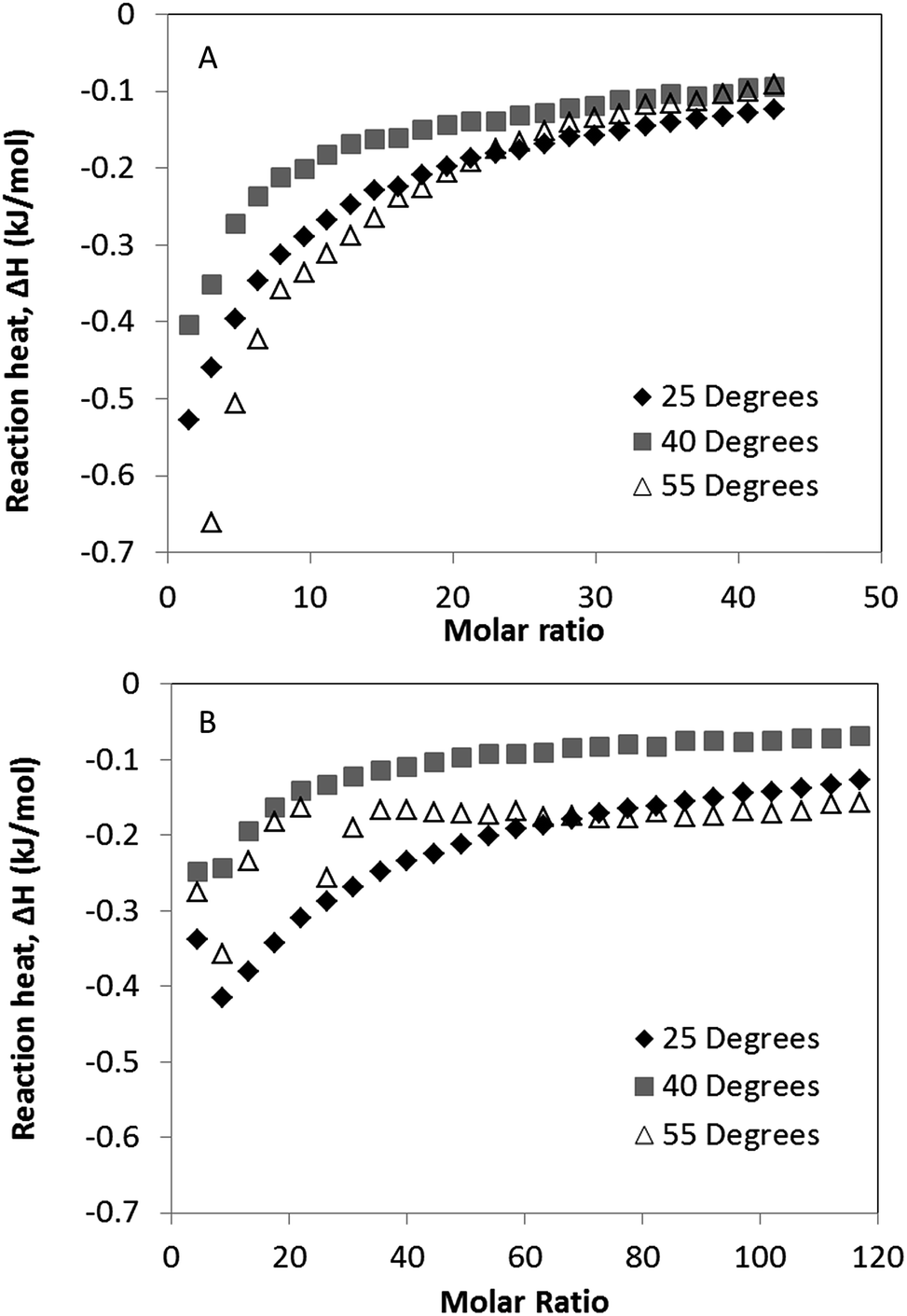 | ||
| Fig. 5 Binding isotherms of polysorbate-20 to both G-CSF (A) and PEG-GCSF (B) between 25 °C and 55 °C. Each point represents the amount of heat released by the protein–surfactant interaction with each titration of polysorbate-20 into the reaction vessel containing 1.43 ml of the protein solution. | ||
:100, and only at high concentrations of surfactant (G-CSF to polysorbate 20 of 1:1000) does the ratio show a deviation from 1, with a value of 0.93, suggesting that the secondary structure of G-CSF slightly decreased at high surfactant concentrations (0.65 wt% polysorbate 20). PEGylated G-CSF shows a similar trend with R values between 1.02 and 1.05 for PEG-GCSF at surfactant ratios ≤1:50, and decreasing to 0.94, only at the highest concentration measured (0.65 wt%), implying that the presence of the PEG chain appended to G-CSF did not alter the structural behaviour of G-CSF initiated by polysorbate 20.
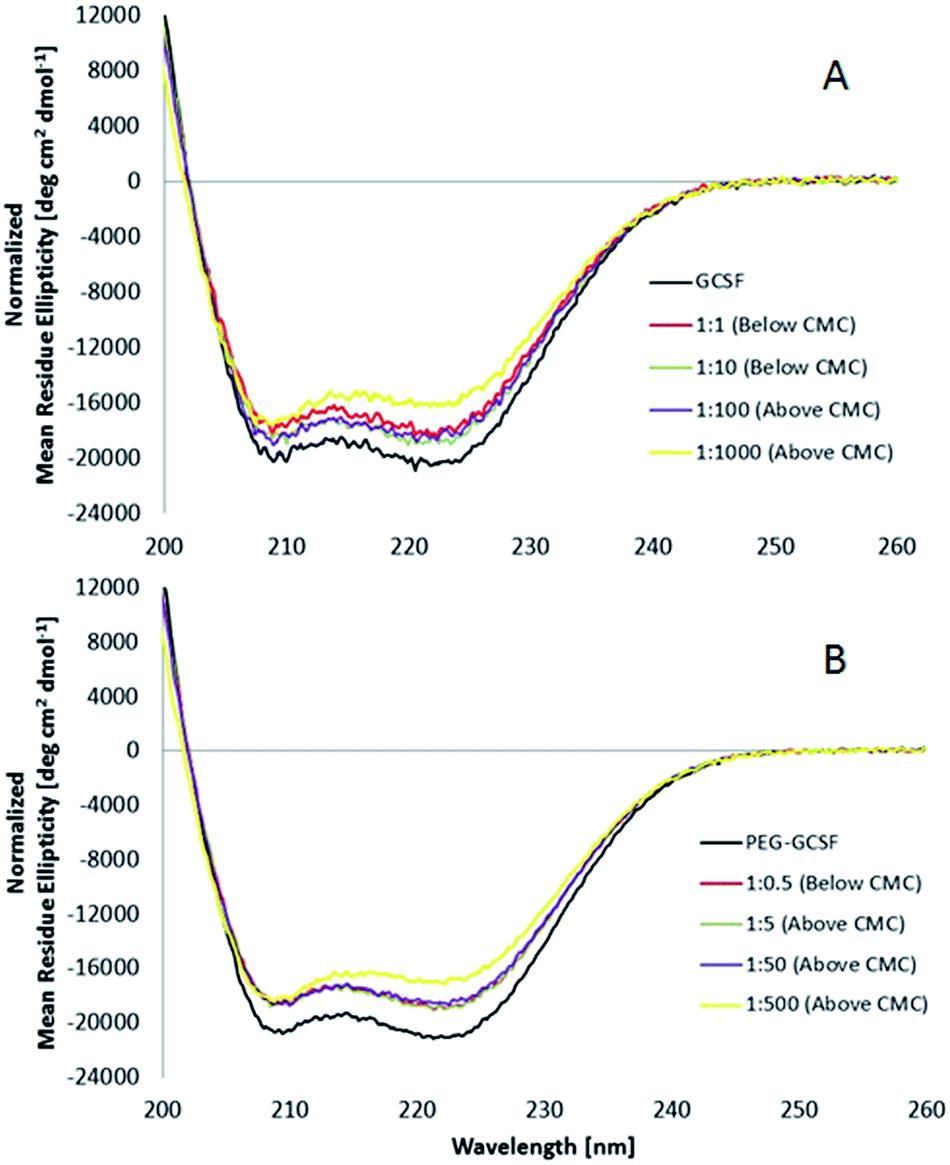 | ||
| Fig. 6 Influence of polysorbate-20 on the circular dichroism spectrum of (A) G-CSF and (B) PEG-GCSF incubated at 25 °C for 12 hours in presence of various concentrations of polysorbate-20, below and above the critical micelle concentration (CMC). Concentrations shown are molar ratio of protein to surfactant concentration. | ||
The θ222/θ208 ratio (R) has also been used to distinguish coiled-coils from single stranded alpha helices. Zhou et al.39 determined empirically that R is equal or higher than 1 for two-stranded coiled-coils, and is lower than 1 (between 0.8 and 0.9) for non-interacting helices. In this study, the θ222/θ208 ratios ranging from 1.01 to 1.05 for G-CSF and PEG-GCSF both alone and in the presence of polysorbate 20 up to concentrations of 0.065 wt%, indicate that the helices of the proteins are associated together in coiled-coils and this 3-dimensional association is not influenced by concentrations of polysorbate 20 up to 0.065 wt%.
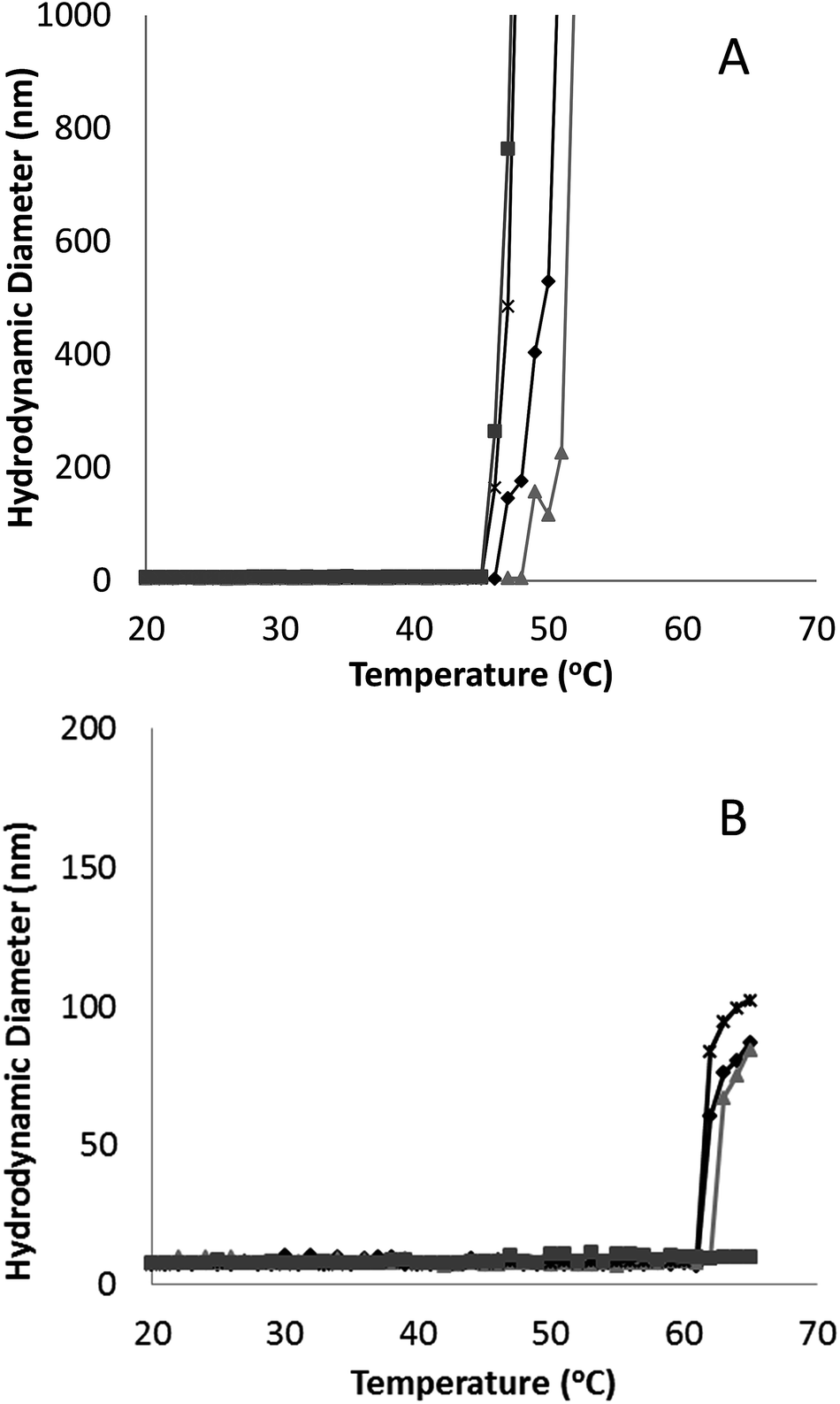 | ||
Fig. 7 Influence of polysorbate 20 concentration on protein aggregation as a function of increasing temperature for (A) G-CSF (1.5 mg ml−1) and (B) PEG-GCSF (1 mg ml−1) in 10 mM acetate buffer at pH 4. The ratio of protein:surfactant are as follows; (A) without polysorbate 20 ( ), 1:0.6 ( ), 1:0.6 ( ), 1:6 ( ), 1:6 ( ), 1:60 ( ), 1:60 ( ) and (B) without polysorbate 20 ( ) and (B) without polysorbate 20 ( ), 1:2 ( ), 1:2 ( ), 1:20 ( ), 1:20 ( ), and 1:200 ( ), and 1:200 ( ). ). | ||
Commercially, protein therapeutics are formulated with protein concentrations of 600 to 1000 μg ml−1 and 10 mg ml−1, respectively for G-CSF and PEG-GCSF.40 Included in the formulation, a concentration of 0.0033 to 0.004 wt% polysorbate 20 is typically used, which is below the cmc (0.007 wt%). This represents approximately one surfactant molecule for every protein molecule for G-CSF, and for PEG-GCSF approximately one surfactant molecule for every ten protein molecules. When combined with the ITC and CD results reported here, it appears that polysorbate 20 interaction with GCSF is weak, does not effect a change in the protein structure that would lead to denaturation, and due to the low ratio of surfactant to protein molecules is possibly transient. PEGylation of GCSF was not found to have influenced the mechanism of aggregation or interaction with polysorbate 20, however did significantly minimise growth in the thermally induced aggregate size, and therefore represents a more physically stable formulation.
4. Conclusion
PEGylation of G-CSF did not alter the three dimensional alpha helical structure or observed temperature of denaturation of the protein. PEGylation of G-CSF did increase the amphiphilic nature, with PEG-GCSF showing greater activity at the buffer–air interface. Both G-CSF and PEG-GCSF demonstrated thermally induced aggregation at temperatures below that of denaturation, and for G-CSF an inverse relationship was observed between concentration and the temperature at which thermal induced aggregation occurred. However, PEGylation did infer significant stability against thermal induced aggregation in solution, increasing the temperature at which aggregation was first observed to approximately 58 °C, from as low as 43 °C for G-CSF, depending on concentration. Conjugation of PEG to G-CSF was shown to limit the thermal induced protein aggregation to a maximum aggregate size of less than 120 nm in diameter, compared to aggregates of greater than 2 μm for G-CSF.In terms of non-ionic surfactant interaction with the proteins, polysorbate 20 did not disturb the alpha helical nature of G-CSF, independent of PEGylation. Direct binding between polysorbate 20 and G-CSF appears to be weak, and to occur through two different mechanisms; one involving individual surfactant molecules and another involving surfactant aggregates. Despite the presence of a hydrated PEG chain, similar binding behaviour was observed between PEG-GCSF and polysorbate 20. These findings increase understanding of stability and interactions in therapeutic protein preparations and may enable more intelligent formulation design.
Acknowledgements
Hopsira Inc. Adelaide is gratefully acknowledged for supplying the G-CSF and PEG-GCSF, for financial support and fruitful discussions.References
- H. J. Lee, A. McAuley, K. F. Schilke and J. McGuire, Adv. Drug Delivery Rev., 2011, 63, 1160–1171 CrossRef CAS PubMed.
- D. S. Pisal, M. P. Kosloski and S. V. Balu-Iyer, J. Pharm. Sci., 2010, 99, 2557–2575 CrossRef CAS PubMed.
- N. B. Bam, J. L. Cleland, J. Yang, M. C. Manning, J. F. Carpenter, R. F. Kelley and T. W. Randolph, J. Pharm. Sci., 1998, 87, 1554–1559 CrossRef CAS PubMed.
- D. K. Chou, R. Krishnamurthy, T. Randolph, J. F. Carpenter and M. C. Manning, J. Pharm. Sci., 2005, 94, 1368–1381 CrossRef CAS PubMed.
- T. Arakawa and Y. Kita, J. Pharm. Sci., 2000, 89, 646 CrossRef CAS PubMed.
- S. Deechongkit, J. Wen, L. O. Narhi, Y. Jiang, S. S. Park, J. Kim and B. A. Kerwin, J. Pharm. Sci., 2009, 98, 3200–3217 CrossRef CAS PubMed.
- D. Otzen, Biochim. Biophys. Acta, Proteins Proteomics, 2011, 1814, 562 CrossRef CAS PubMed.
- N. B. Bam, T. W. Randolph and J. L. Cleland, Pharm. Res., 1995, 12, 2–11 CrossRef CAS.
- T. W. Randolf and L. S. Jones, in Pharm. Biotechnol., ed. C. J. F. and M. M. C., Kluwer Academic/Plenum Publishers, NewYork, 2002, vol. 13, pp. 159–175 Search PubMed.
- A. Abuchowski, J. R. McCoy, N. C. Palczuk, T. van Es and F. F. Davis, J. Biol. Chem., 1977, 252, 3582 CAS.
- G. Molineux, Cancer Treat. Rev., 2002, 28, 13 CrossRef CAS PubMed.
- B. Plesner, C. J. Fee, P. Westh and A. D. Nielson, Eur. J. Pharm. Biopharm., 2011, 79, 399–405 CrossRef CAS PubMed.
- A. Natalello, D. Ami, M. Collini, L. D'Alfons, G. Chirico, G. Tonon, S. Scaramuzza, R. Schrepfer and S. M. Doglia, PLoS One, 2012, 7, e42511 CAS.
- R. S. Rajan, T. Li, M. Aras, C. Sloey, W. Sutherland, H. Arai, R. Briddell, O. Kinstler, A. M. K. Lueras, Y. Zhang, H. Yeghnazar, M. Treuheit and D. N. Brems, Protein Sci., 2006, 15, 1063–1075 CrossRef CAS PubMed.
- A. Grigoletto, A. Mero, I. Zanusso, O. Schiavon and G. Pasut, Macromol. Biosci., 2016, 16, 50–60 CrossRef CAS PubMed.
- L. S. Holm, A. Mcumber, J. E. Rasmussen, M. Obiols-Rabasa, P. W. Thulstrup, M. R. Kasimova, T. W. Randolph and M. van de Weert, J. Pharm. Sci., 2014, 103, 3043–3054 CrossRef CAS PubMed.
- G. D. Demetri and J. D. Griffin, Blood, 1991, 78, 2791 CAS.
- D. Metcalf, Blood, 1986, 67, 257–267 CAS.
- I. A. Tabbara, South. Med. J., 1993, 86, 350 CrossRef CAS PubMed.
- K. Yamazaki, T. Iwura, K. Murayama, R. Ishikawa and Y. Ozaki, Vib. Spectrosc., 2005, 38, 33–38 CrossRef CAS.
- T. C. Souza, T. C. Boone, J. Gabrilove, P. H. Lai, K. M. Zsebo, D. C. Murdock, V. R. Chazin, J. Bruszewski, H. Lu and K. K. Chen, Science, 1986, 232, 61–65 Search PubMed.
- M. J. Treuheit, A. A. Kosky and D. N. Brems, Pharm. Res., 2002, 19, 511–516 CrossRef CAS.
- J. W. Regini, H. Ecroyd, S. Meehan, K. Bremmell, M. J. Clarke, D. Lammie, T. Wess and J. A. Carver, Mol. Vision, 2010, 16, 2446–2456 CAS.
- M. M. Pierce, C. Raman and B. T. Nall, Methods, 1999, 19, 213–221 CrossRef CAS PubMed.
- L. S. Mizoue and J. Tellinghuisen, Anal. Biochem., 2004, 326, 125–127 CrossRef CAS PubMed.
- S. W. Raso, J. Abel, J. M. Barnes, K. M. Maloney, G. Pipes, M. J. Treuheit, J. King and D. N. Brems, Protein Sci., 2005, 14, 2246–2257 CrossRef CAS PubMed.
- R. W. Niven, S. J. Prestrelski, M. J. Treuheit, A. Y. Ip and T. Arakawa, Int. J. Pharm., 1996, 127, 191–201 CrossRef CAS.
- C. Pinholt, J. T. Bukrinsky, S. Hostrup, S. Frokjaer, W. Norde and L. Jorgensen, Eur. J. Pharm. Biopharm., 2011, 77, 139–147 CrossRef CAS PubMed.
- F. He, S. Hogan, R. F. Latypov, L. O. Narhi and V. I. Razinkov, J. Pharm. Sci., 2010, 99, 1707–1720 CrossRef CAS PubMed.
- T. Menzen and W. Friess, J. Pharm. Sci., 2013, 102, 415–428 CrossRef CAS PubMed.
- I. M. Kempson, P. Chang, K. Bremmell and C. A. Prestidge, Langmuir, 2013, 29, 15573–15578 CrossRef CAS PubMed.
- H. L. Kim, A. McAuley and J. McGuire, J. Pharm. Sci., 2014, 103, 1337 CrossRef CAS PubMed.
- B. L. Osborn, L. Sekut, M. Corcoran, C. Poortman, B. Sturm, G. Chen, D. Mather, H. L. Lin and T. J. Parry, Eur. J. Pharmacol., 2002, 456, 149–158 CrossRef CAS PubMed.
- A. D. Nielsen, K. Borch and P. Westh, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 2000, 1479, 321–331 CrossRef CAS.
- S. Salmaso, S. Bersani, F. Mastrotto, G. Tonon, R. Schrepfer, S. Genovese and P. Caliceti, J. Controlled Release, 2012, 162, 176–184 CrossRef CAS PubMed.
- L. S. Holm, P. W. Thulstrup, M. R. Kasimova and M. van de Weert, PLoS One, 2015, 10, e0133584 Search PubMed.
- C. Hoffmann, A. Blume, I. Miller and P. Garidel, Eur. Biophys. J., 2009, 38, 557–568 CrossRef CAS PubMed.
- T. S. Banipal, A. Kaur, I. A. Khan and P. K. Banipal, RSC Adv., 2016, 6, 34754–34769 RSC.
- N. E. Zhou, C. M. Kay and R. S. Hodges, J. Biol. Chem., 1992, 267, 2664–2670 CAS.
- MIMS Australia, MIMs Online, 2016.
| This journal is © The Royal Society of Chemistry 2016 |