
Altenusin derivatives from mangrove endophytic fungus Alternaria sp. SK6YW3L†
Abstract
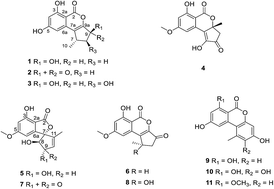
Five new altenusin derivatives, compounds 1–5, along with six known analogues 6–11, were isolated from a culture of the endophytic fungus Alternaria sp. SK6YW3L, which was isolated from a fresh fruit of the mangrove plant Sonneratia caseolaris, collected from the South China Sea. Their structures were elucidated by analysis of 1D and 2D NMR and high resolution mass spectroscopic data. The absolute configurations of compounds 1–3 and 5 were assigned by quantum chemical calculations of the electronic circular dichroic (ECD) spectra. Structures of compounds 1 and 4 were further confirmed by single-crystal X-ray diffraction experiments using Cu Kα radiation. All isolated compounds were evaluated for α-glucosidase inhibitory activity, and compounds 2, 3 and 9 exhibited moderate inhibitory activity. The plausible biosynthetic pathways for all the compounds were proposed.
 Please wait while we load your content...
Please wait while we load your content...

