
Direct oxidative amination of aromatic aldehydes with amines in a continuous flow system using a metal-free catalyst†
Kai
Guo  *ac
*ac
*ac
Abstract
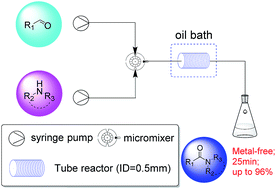
A novel method for metal-free oxidative amination of aromatic aldehydes and alcohols in the presence of H2O2/NaBr/H+ within 25 min in a continuous flow system has been developed. A series of different substrates were tested and the corresponding products were obtained in good yields.
 Please wait while we load your content...
Please wait while we load your content...

