DOI:
10.1039/C6RA16113H
(Communication)
RSC Adv., 2016,
6, 75032-75037
Carbon dioxide affects the phase transition of poly(N-isopropylacrylamide)†
Received
21st June 2016
, Accepted 3rd August 2016
First published on 4th August 2016
Abstract
The effects of atmospheres of CO2 and N2 on the lower critical solution temperature (LCST) of poly(N-isopropylacrylamide) (PNIPAAm) in aqueous solution have been investigated using high-pressure differential scanning calorimetry (HP-DSC). In the absence of CO2 and N2, the phase transition—from the hydrated to dehydrated state—of PNIPAAm in aqueous solution was characterized by an endothermic peak near 30.5 °C, namely the LCST. This endothermic peak shifted to relatively lower temperature upon increasing the pressure of CO2, but shifted slightly to higher temperature under a higher pressure of N2. This behavior appears to be associated with the CO2 molecules displacing the H2O molecules from around the amide groups of PNIPAAm upon increasing the pressure, thereby enhancing the formation of intramolecular hydrogen bonds between the amide groups; in contrast, increasing the N2 pressure strengthened the interactions of H2O with the apolar isopropyl and amide groups. Despite the difference in the effects of CO2 and N2 on the LCST, higher pressures of CO2 and N2 both led to more positive changes in enthalpy (ΔH) for the phase transition per mole of NIPAAm units. The higher values of ΔH at higher CO2 pressures presumably resulted from the formation of strong intramolecular hydrogen bonds. For a given CO2 pressure, the value of ΔH was less positive at higher concentrations of PNIPAAm, suggesting a lesser degree of disruption of hydrogen bonds during its HP-DSC heating scans. Under a given pressure of CO2, the addition of a salt (NaCl, KCl, KBr) led to a further decrease in the LCST and the value of ΔH of the aqueous PNIPAAm solution, due to the salt ions coordinating H2O molecules.
Introduction
Poly(N-isopropylacrylamide) (PNIPAAm) is soluble in water and adopts a coil conformation at relatively low temperature, whereas it is insoluble and has a globule conformation at temperatures higher than its lower critical solution temperature (LCST, ca. 32 °C).1–5 The coil–globule phase transition of PNIPAAm in aqueous solution at the LCST is an endothermic process that can be observed in DSC heating curves;6,7 it results from dehydration of ordered H2O molecules around the polar amide and apolar isopropyl groups. The Maeda8 and Sun9 groups used Fourier transform infrared (FTIR) spectroscopic analyses to investigate the dehydration of the isopropyl groups at higher temperatures, observing the shift to lower wavenumbers of the asymmetric C–H stretching band of the isopropyl groups. Maeda et al.8 also found that dehydration of the polar amide groups occurred at temperatures above the LCST, but most of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O functionalities (ca. 87%) in the amide groups of PNIPAAm in the globule state remained hydrogen-bonded with H2O molecules while the others (ca. 13%) formed intra- or interchain hydrogen bonds with the N–H units of PNIPAAm. The addition of metal halide salts (e.g., NaCl, KCl, KBr, KI) can expedite the dehydration, thereby lowering the LCST of PNIPAAm in aqueous solutions.8,10 These hygroscopic substances are commonly used as salting-out agents to separate less-hydrophilic molecules from H2O.
O functionalities (ca. 87%) in the amide groups of PNIPAAm in the globule state remained hydrogen-bonded with H2O molecules while the others (ca. 13%) formed intra- or interchain hydrogen bonds with the N–H units of PNIPAAm. The addition of metal halide salts (e.g., NaCl, KCl, KBr, KI) can expedite the dehydration, thereby lowering the LCST of PNIPAAm in aqueous solutions.8,10 These hygroscopic substances are commonly used as salting-out agents to separate less-hydrophilic molecules from H2O.
The CO groups in polymers can interact specifically with CO2 molecules, leading to much more significant dissolution of CO2 in these polymers than that of N2, which does not interact with CO groups.11–13 The presence of CO2 around the CO groups of PNIPAAm in aqueous solutions might, therefore, displace H2O molecules from around the apolar isopropyl groups and/or the polar amide groups, thereby decreasing the LCST of PNIPAAm. In this study, we used high-pressure differential scanning calorimetry (HP DSC) to monitor the effect of CO2 on the LCST behavior of PNIPAAm aqueous solutions with respect to the CO2 pressure. For comparison, we also examined the effects of the N2 pressure and hygroscopic salts (metal halides) on the phase transitions of PNIPAAm aqueous solutions.
Experimental section
PNIPAAm
The PNIPAAm polymer was prepared through free radical polymerization of NIPAAm monomer (Tokyo Kasei) using azobis(isobutyronitrile) (AIBN, Aldrich) as the free radical initiator. To prepare low-molecular-weight PNIPAAm, the purified NIPAAm monomer and AIBN (molar ratio: 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) were dissolved in MeOH and heated at 55 °C for 2 h under a N2 atmosphere; the total concentration of NIPAAm and AIBN was 2.2 M. To prepare high-molecular-weight PNIPAAm, a homogeneous reactant mixture (NIPAAm/AIBN molar ratio: 40:1) was loaded in an 8 mL vial and then placed in a 200 mL high-pressure vessel. CO2 was loaded into the high-pressure vessel and the free radical polymerization of NIPAAm was conducted under a CO2 pressure of 27.6 MPa at 55 °C for 8 h. After polymerization, the vessel was depressurized and the settled product inside the vial was subjected to purification as described previously.14 Using an Ubbelohde viscometer, the intrinsic viscosity ([η]) was measured; the molecular weight was calculated using the equation15
:1) were dissolved in MeOH and heated at 55 °C for 2 h under a N2 atmosphere; the total concentration of NIPAAm and AIBN was 2.2 M. To prepare high-molecular-weight PNIPAAm, a homogeneous reactant mixture (NIPAAm/AIBN molar ratio: 40:1) was loaded in an 8 mL vial and then placed in a 200 mL high-pressure vessel. CO2 was loaded into the high-pressure vessel and the free radical polymerization of NIPAAm was conducted under a CO2 pressure of 27.6 MPa at 55 °C for 8 h. After polymerization, the vessel was depressurized and the settled product inside the vial was subjected to purification as described previously.14 Using an Ubbelohde viscometer, the intrinsic viscosity ([η]) was measured; the molecular weight was calculated using the equation15
The calculated molecular weights of the low- and high-molecular-weight PNIPAAm samples were 3.90 × 105 and 1.13 × 106 g mol−1, respectively.
HP-DSC measurements of phase transitions of aqueous PNIPAAm solutions
Certain amounts of PNIPAAm were dissolved in deionized water to prepare aqueous PNIPAAm solutions having concentrations of 1.5, 5, and 10 wt%. A portion (ca. 5 mg) of the aqueous solution was taken (micropipette) and placed into an aluminum sample pan and weighed; the pan loaded without a lid was into the heating chamber of a HP-DSC apparatus (HP DSC-Q20p, TA Instruments). The chamber was then purged with CO2 for 5 min before heating under CO2 at pressures of 0.69, 1.38, 2.07, 2.76, 3.45, and 4.14 MPa while increasing the temperature from 20 to 40 °C at a rate of 2 °C min−1. For comparison, the HP-DSC apparatus was also purged with N2 for 5 min before heating under N2 at pressures of 1.38 and 2.76 MPa while increasing the temperature from 20 to 40 °C at a rate of 2 °C min−1. The peak temperature and area of the endothermic peak in the first heating curve of each sample were recorded and taken to be the LCST and enthalpy, respectively, of the phase transition of the PNIPAAm aqueous solution. A 5 wt% PNIPAAm aqueous solution containing 1 wt% of a salt (KBr, KCl, or NaCl) was also heated under CO2 at various pressures in the HP-DSC apparatus to investigate the effects of salts on the phase transitions of aqueous PNIPAAm solutions.
Results and discussion
Effects of CO2 and N2 on LCST behavior of aqueous PNIPAAm solutions
Fig. 1 and 2 present the HP-DSC first heating curves for aqueous PNIPAAm solutions of 1.5, 5, and 10 wt% in CO2 and N2, respectively, at various pressures. An endothermic peak appears in every heating scan, corresponding to the coil-to-globule phase transition16 of the aqueous PNIPAAm solution from the dissolved state to the turbid state.6,7 For PNIPAAm solutions with different concentrations without CO2, the peaks were ca. 30.5 °C and the peak in Fig. 1(A)–(C) shifted to lower temperatures upon increasing the CO2 pressure; in contrast, it remained almost unchanged upon increasing the pressure of N2, as revealed in Fig. 2(A)–(C). In other words, the LCST of aqueous PNIPAAm solutions could decrease upon increasing the CO2 pressure, but slightly increase upon increasing the N2 pressure.
 |
| | Fig. 1 (A–C) HP-DSC first-heating curves (2 °C min−1) of (A) 1.5, (B) 5, and (C) 10 wt% aqueous PNIPAAm solutions under CO2 at pressures of (a) 0, (b) 0.69, (c) 1.38, (d) 2.07, (e) 2.76, (f) 3.45, and (g) 4.14 MPa. (D) Summary of the corresponding LCSTs and values of ΔH. | |
 |
| | Fig. 2 (A–C) HP-DSC first-heating curves (2 °C min−1) of (A) 1.5, (B) 5, and (C) 10 wt% aqueous PNIPAAm solutions under N2 at pressures of (a) 0, (b) 1.38, and (c) 2.76 MPa. (D) Summary of the corresponding LCSTs and values of ΔH. | |
Because the CO groups in polymers can interact specifically with CO2,11 but not with N2, the dissolution of CO2 in such polymers can be much greater than that of N2.12,13 Fig. 1(D) reveals that the LCSTs of the aqueous PNIPAAm solutions clearly decreased upon increasing the pressure of CO2 at various concentrations. In contrast, the LCST increased slightly upon increasing the N2 pressures, as displayed in Fig. 2(D). This behavior can be attributed to CO2, but not N2, displacing the H2O molecules around the apolar isopropyl groups and/or the polar amide groups in PNIPAAm. Upon increasing the CO2 pressure, the displacement of H2O molecules increased and the formation of intramolecular hydrogen bonds between the amide groups in PNIPAAm was, thereby, promoted, leading to a decrease in its LCST. Upon increasing the N2 pressure, the increase in the LCST might be attributable to the pressure effect of N2: enhancing the interactions of H2O with the apolar isopropyl and polar amide groups of PNIPAAm and, thereby, promoted the solvation and/or hydrogen bonding of these groups with H2O, leading to an increase in the LCST. Scheme 1 summarizes the possible interactions of PNIPAAm in aqueous solutions under N2 and CO2.
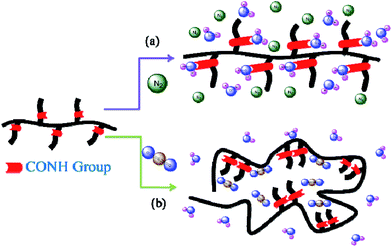 |
| | Scheme 1 Cartoon representation of the possible interactions of PNIPAAm under (a) N2 and (b) CO2 atmospheres. | |
Effects of CO2 and N2 on value of ΔH of phase transition of PNIPAAm solution
Fig. 1(D) and 2(D) and Table S1† summarize the enthalpy changes (ΔH) and LCST behavior of the coil-to-globule phase transitions of aqueous PNIPAAm solutions at three different concentrations, as measured through HP-DSC under various pressures of CO2 and N2, respectively. The coil-to-globule phase transition of PNIPAAm in H2O upon heating is an endothermic process resulting from disruption of the interactions of H2O with the apolar isopropyl groups and the hydrogen bonds of H2O with the polar amide groups. Despite the difference in the effects of the pressure of CO2 and N2 on their LCST as discussed above, relatively high pressures of CO2 and N2 both led to more positive values of ΔH for the phase transition per mole of NIPAAm units. Because the LCST of the aqueous PNIPAAm solution decreased upon increasing the pressure of CO2, we were surprised that a higher CO2 pressure provided a more positive value of ΔH for the phase transition of PNIPAAm. This finding suggests that although the displacement of H2O molecules by CO2 molecules led to a decrease in LCST, the formation of intramolecular hydrogen bonds among the amide groups was promoted under CO2, thereby increasing the value of ΔH upon increasing the CO2 pressure. For a given pressure of CO2 or N2, the endothermic value of ΔH was lower at higher concentrations of PNIPAAm, as is evident in Fig. 1(d) and 2(d). This behavior might be associated with less disruption of the hydrogen bonds during the heating of PNIPAAm at higher concentrations.
Effect of salts on LCST and value of ΔH of phase transition of PNIPAAm solution
NaCl, KCl, and KBr are hygroscopic salts that can be used as salting-out agents to separate less-hydrophilic molecules from H2O. They are soluble in H2O and dissociate into their constituent anions and cations, which interact strongly with the positive and negative ends, respectively, of H2O dipoles, leading to hygroscopicity in air and rapid dissolution in H2O. Fig. 3 displays the variations in the LCSTs of 5 wt% PNIPAAm solutions containing 1 wt% of KBr, KCl, or NaCl, plotted with respect to the pressure of CO2. The LCST of each sample decreased upon the addition of each salt at each CO2 pressure. This behavior is consistent with these salts coordinating to H2O molecules and, thereby, decreasing the solvation power of H2O molecules to the apolar isopropyl groups and the hydrogen bonding of H2O molecules to the polar amide groups of PNIPAAm. In the absence of CO2, the solvation power and/or hydrogen bonding of H2O decreased the most in the presence of NaCl, followed by KCl and, to the least extent, KBr. This sequence suggests that NaCl absorbs H2O the most and KBr the least, with KCl in between the two extremes. A Na+ cation is smaller than a K+ cation, and a Cl− anion is smaller than a Br− anion. Because the weight concentration of each added salt was 1 wt%, a smaller cation or anion would give a higher molar concentration of the dissociated ions and, thus, a higher ionic strength, resulting in greater absorption of H2O molecules.
 |
| | Fig. 3 LCSTs and values of ΔH for the phase transitions of 5 wt% aqueous PNIPAAm solutions containing 1 wt% of a salt (KBr, KCl, or NaCl), determined from HP-DSC first-heating scans (2 °C min−1) under CO2 at various pressures. | |
Fig. 3 and Table S2† also summary the values of ΔH for the phase transitions of aqueous PNIPAAm solutions in the presence of these salts. For a given pressure of CO2, the endothermic value of ΔH decreased upon the addition of a salt. NaCl, with its greatest ionic strength, caused the greatest decrease in the value of ΔH, while KBr, of lowest ionic strength, induced the lowest decrease. Upon the addition of a salt, the decrease in the value of ΔH for the phase transition of PNIPAAm in aqueous solution indicated greater disruption of the interactions/hydrogen bonds of H2O with the apolar isopropyl groups and the polar amide groups of PNIPAAm, suggesting that the H2O molecules around the apolar isopropyl groups and the polar amide groups had been absorbed by the added salt. Thus, the decreases in the LCST of PNIPAAm induced by CO2 or the salts arose through different mechanisms: the CO2 molecules displacing H2O molecules and the salts absorbing H2O molecules from around the apolar isopropyl groups and the polar amide groups of PNIPAAm.17,18
Effect of molecular weight of PNIPAAm on CO2-dependent LCST
Two PNIPAAm polymers having different weight-average molecular weights (Mw) were used to investigate the effect of molecular weight on the CO2-dependent LCST and values of ΔH as shown in Table S3.† Fig. 4 reveals that, for both polymers, the LCST decreased and the value of ΔH increased upon increasing the pressure of CO2. The higher-Mw polymer provided the lower LCST at any CO2 pressure observed through HP-DSC. The lower LCST for the higher-Mw PNIPAAm may have arisen from greater entanglement, more intrachain hydrogen bonds, and more hydrophobic interactions; similar phenomena have been reported previously.19–25 The greater degree of intrachain hydrogen bonding and hydrophobic interactions in the higher-Mw PNIPAAm should lead to less intermolecular hydrogen bonding between PNIPAAm and H2O at any CO2 pressure; if so, less heat would be required to disrupt the intermolecular hydrogen bonds during the phase transition at the LCST. Indeed, the enthalpy change (ΔH = 3.44 kJ mol−1) for the phase transition at the LCST for the higher-Mw PNIPAAm was lower than that (ΔH = 4.09 kJ mol−1) for the lower-Mw PNIPAAm.
 |
| | Fig. 4 LCSTs and values of ΔH for the phase transitions of 5 wt% aqueous PNIPAAm solutions [weight-average molecular weights: 0.39 × 106 (low Mw) and 1.13 × 106 (high Mw) g mol−1], determined from HP-DSC first-heating scans (2 °C min−1) under CO2 at various pressures. | |
Conclusions
The interactions of CO2 with the CO units of PNIPAAm can displace H2O molecules from around the amide groups of PNIPAAm in aqueous solution leading to a decrease in the LCST of the polymer. In contrast, under N2, which does not interact with the CO units of PNIPAAm, the H2O molecules can interact more strongly with the apolar isopropyl and polar amide groups of PNIPAAm in aqueous solution, resulting in an increase in the LCST of the polymer. Despite the difference in the effects of CO2 and N2 on the shifting LCST, higher pressures of CO2 and N2 both led to increases in the endothermic value of ΔH for the phase transition of PNIPAAm. The addition of salts (NaCl, KCl, KBr) further decreased the LCST of PNIPAAm in aqueous solution at any CO2 pressure, due to the absorption of H2O by the salts in addition to the displacement of H2O molecules by CO2 molecules.
Acknowledgements
We thank the Ministry of Science and Technology of Taiwan for supporting this study financially under contract MOST 103-2221-E-390-030.
References
- C. T. Lai, R. H. Chien, S. W. Kuo and J. L. Hong, Macromolecules, 2011, 44, 6546–6556 CrossRef CAS.
- Y. W. Lai, S. W. Kuo and J. L. Hong, Langmuir, 2012, 28, 15725–15735 CrossRef PubMed.
- C. W. Tu and S. W. Kuo, J. Polym. Res., 2014, 21, 476 CrossRef.
- K. Liu, P. Pan and Y. Bao, RSC Adv., 2015, 5, 94582–94590 RSC.
- J. Gan, X. X. Guan, J. Zheng, H. Guo, K. Wu, L. Liang and M. Lu, RSC Adv., 2016, 6, 32967–32978 RSC.
- C. Boutris, E. G. Chatzi and C. Kiparissides, Polymer, 1997, 38, 2567–2570 CrossRef CAS.
- J. Zhang and N. A. Peppas, Macromolecules, 2000, 33, 102–107 CrossRef CAS.
- Y. Maeda, T. Higuchi and I. Ikeda, Langmuir, 2000, 16, 7503–7509 CrossRef CAS.
- B. J. Sun, Y. N. Lin, P. Y. Wu and H. W. Siesler, Macromolecules, 2008, 41, 1512–1520 CrossRef CAS.
- Y. W. Lai, S. W. Kuo and J. L. Hong, RSC Adv., 2012, 2, 8194–8200 RSC.
- S. G. Kazarian, M. F. Vincent, F. V. Bright and C. L. Liotta, J. Am. Chem. Soc., 1996, 118, 1729–1736 CrossRef CAS.
- Y. T. Shieh and K. H. Liu, J. Supercrit. Fluids, 2003, 25, 261–268 CrossRef CAS.
- Y. T. Shieh and K. H. Liu, J. Polym. Res., 2002, 9, 107–113 CrossRef CAS.
- Y. T. Shieh, C. Zhao, T. L. Wang and C. H. Yang, J. Supercrit. Fluids, 2014, 91, 1–6 CrossRef CAS.
- O. Chiantore, M. Guaita and L. Trossarelli, Makromol. Chem., 1979, 180, 969–973 CrossRef CAS.
- C. Wu and X. Wang, Phys. Rev. Lett., 1998, 80, 4092–4094 CrossRef CAS.
- J. Spevacek, J. Dybal, L. Starovoytova, A. Zhigunov and Z. Sedlakova, Soft Matter, 2012, 8, 6110–6119 RSC.
- J. Spevacek, R. Konefal and E. Vadova, Macromol. Chem. Phys., 2016, 217, 1370–1375 CrossRef CAS.
- H. G. Schild and D. A. Tirrell, J. Phys. Chem., 1990, 94, 4352–4356 CrossRef CAS.
- T. Baltes, F. Garret-Flaudy and R. Freitag, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2977–2989 CrossRef CAS.
- S. Furyk, Y. Zhang, D. Ortiz-Acosta, P. S. Cremer and D. E. Bergbreiter, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 1492–1501 CrossRef CAS.
- Y. T. Shieh and B. H. Chen, J. Supercrit. Fluids, 2016, 107, 624–629 CrossRef CAS.
- V. O. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2006, 196, 1–85 CrossRef CAS.
- V. O. Aseyev, H. Tenhu and F. M. Winnik, Adv. Polym. Sci., 2016, 242, 29–89 CrossRef.
- F. Messussen, E. Nies, H. Berghmans, S. Verbrugghe, E. Goethals and F. Du-Prez, Polymer, 2000, 41, 8597–8602 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra16113h |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 




