DOI:
10.1039/C6RA16050F
(Paper)
RSC Adv., 2016,
6, 75578-75587
A kinetic and mechanistic study of the degradation of 1,2-dichloroethane and methyl tert-butyl ether using alkaline-activated persulfate oxidation
Received
21st June 2016
, Accepted 29th July 2016
First published on 29th July 2016
Abstract
In this study, alkaline-activated persulfate is used to treat groundwater that is contaminated with 1,2-dichloroethane (1,2-DCA)- and methyl tert-butyl ether (MTBE). The results show that alkaline-activated persulfate accelerates the degradation of 1,2-DCA. Vinyl chloride is produced because there is dehydrochlorination of 1,2-DCA at high pH. Although the rate of degradation of MTBE in alkaline-activated persulfate systems is decreased, there is a significant reduction in the accumulation of MTBE-degrading byproducts, tert-butyl alcohol and tert-butyl formate. Alkaline-activated persulfate degrades 1,2-DCA via the mechanisms of oxidation and dehydrochlorination, but oxidation alone contributes to the removal of MTBE. The use of low-cost basic oxygen furnace (BOF) slag increases the pH of the solution (>12) and allows the alkaline-activated persulfate reaction to proceed. No detectable heavy metals are released from the BOF slag in the persulfate system that is activated by BOF slag. The degradation of 1,2-DCA and MTBE follows the pseudo-first-order kinetics for all persulfate systems. Alkaline-activated persulfate causes a decrease in ORP because there is a high pH, which adversely affects the removal of contaminants if the contaminants do not undergo base-mediated degradation. Therefore, the performance in terms of contaminant removal must be evaluated before alkaline-activated persulfate is used for different compounds. This novel, slag-activated persulfate system is an environmentally friendly and cost-effective alternative to the remediation of groundwater.
1. Introduction
Groundwater contamination is a global issue because of the potential threat of toxic pollutants to water resources and human health. Generally, petroleum and chlorinated hydrocarbons are the most commonly detected pollutants in groundwater.1–4 1,2-Dichloroethane (DCA), the most commonly produced halogenated chemical in the world, is mainly used in the synthesis of vinyl chloride (VC), which is used for the production of polyvinyl chloride. 1,2-DCA has been classified as a probable human carcinogen (Group B2) by the US Environmental Protection Agency (US EPA)5 and a possible human carcinogen (Group 2B) by the International Agency for Research on Cancer (IARC).6 The US EPA has set a maximum contaminant level (MCL) for 1,2-DCA in drinking water at 0.005 mg L−1, to protect public health.7 Methyl tert-butyl ether (MTBE) is the most commonly used gasoline additive because of its low cost and ease of production and blending.8 Since MTBE is a potential human carcinogen at high doses, the US EPA has set an advisory level for drinking water of 20–40 μg L−1.9 There is significant potential for groundwater contamination by 1,2-DCA and MTBE because of their widespread use. Both 1,2-DCA and MTBE are also resistant to biodegradation and chemical oxidation, which renders groundwater remediation difficult.10–13
In situ chemical oxidation (ISCO) is a rapid and effective remediation technology that uses powerful oxidants to treat high levels of contaminants in the subsurface.14 Hydrogen peroxide, permanganate and persulfate are common oxidants that are used in aqueous form for ISCO. Persulfate oxidation has been developed to remediate contaminated soil and groundwater. Persulfate is a strong and relatively stable oxidant that is activated by heat (reaction (1))15,16 or a transition metal (Mn+) (reaction (2))17,18 to produce more powerful sulfate free radicals that degrade a wide range of organic pollutants:
| | |
S2O82− + heat → 2SO4−˙
| (1) |
| | |
S2O82− + Mn+ → SO4−˙ + SO42− + Mn+1
| (2) |
| | |
SO4−˙ + e− → SO42−, E0 = 2.6 V
| (3) |
In addition to heat and transition metals, many other activation technologies, such as chelated Fe(II),19,20 zero valent iron (ZVI),21,22 ultraviolet light23,24 and activated carbon25 are also used to activate persulfate. Recently, alkaline-activated persulfate (alkaline persulfate) has been used for groundwater remediation.26–28 The notable advantage of alkaline-activated persulfate is that the addition of a base neutralizes the acid that is generated from persulfate during the treatment.29 Since a decrease in pH can result in the dissolution of heavy metals in soil,14 alkaline persulfate also reduces the possibility of heavy metal contamination during site remediation.
In general, a pH of greater than 10 is required to activate persulfate.26,27,30,31 At a high pH, sulfate radicals react with hydroxyl ions and water to generate hydroxyl radicals:27,32
| | |
SO4−˙ + OH− → ˙OH + SO42−
| (4) |
| | |
SO4−˙ + H2O → ˙OH + SO42− + H+
| (5) |
Two possible mechanisms for alkaline-activated persulfate have been proposed. Corbin III30 reported that under alkaline conditions, sulfate radicals convert hydroxyl ions to hydroxyl radicals (reaction (4)) and then propagation reactions generate other reactive species:
| | |
S2O82− + 2H2O → 2HSO4− + H2O2
| (7) |
| | |
H2O2 + OH− → HO2− + H2O
| (8) |
| | |
H2O2 + HO2− → O2˙− + ˙OH + H2O
| (9) |
Furman et al.33 noted that the first step in the alkaline activation of persulfate is the base-catalyzed hydrolysis of persulfate to form peroxomonosulfate (SO52−) and sulfate and then other reactions occur:
| |
 | (10) |
| |
 | (11) |
| |
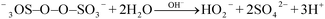 | (12) |
| | |
H–O–O− + −3OS–O–O–SO3− → SO4˙− + SO42− + H+ + O2˙−
| (13) |
| | |
2S2O82− + 2H2O → 3SO32− + SO4˙− + O2˙− + 4H+
| (14) |
The sulfate radicals that are produced in reactions (13) and (14) then react with hydroxyl ions to generate hydroxyl radicals, as shown in reaction (4).
Alkaline-activated persulfate has been successfully demonstrated to increase the removal of different contaminants. Block et al.26 reported that alkaline persulfate (pH > 10) is an aggressive oxidation system that can completely remove problematic compounds, such as chlorinated methanes and ethanes. Liang et al.34 measured trichloroethylene (TCE) removal by persulfate using ethylenediamine-tetra-acetic acid (EDTA) chelated Fe3+ as an activator at different pH values (pH 3, 7 and 10). The results showed that the rate at which TCE is removed increases as the pH value increases, because hydroxyl radicals are generated in the system (reaction (4)). Nitrobenzene is used to identify sulfate and hydroxyl radicals in thermally activated persulfate under different pH conditions.35 The results show that nitrobenzene degradation rates are greater at pH values of 12 and 9 than at pH values of 7, 4 and 2. This study showed that predominant hydroxyl radicals are generated in basic solutions.
Although powerful hydroxyl radicals occur in alkaline persulfate systems, many studies have shown that alkaline-activated persulfate retards the degradation of pollutants. The rate at which MTBE is degraded by thermally activated persulfate decreases as the pH value increases (pH 2.5–12) because sulfate radicals and hydroxyl radicals react rapidly with hydroxyl ions under high pH conditions.36 Huling et al.37 found that MTBE degradation is better under neutral and acid conditions (pH 3.5–7.5) than under alkaline conditions (pH 11–12.5). Sulfate radicals are scavenged by water and hydroxyl ions and the hydroxyl radicals are consumed by sulfate ions and other reactants in the system. The effect of different persulfate processes, such as heating, Fe(III)–EDTA, alkaline activation and hydrogen peroxide-persulfate binary mixtures, on the degradation of organic pollutants has been studied. The results show that alkaline-activated persulfate is the least efficient way to degrade pollutants.29,38
For alkaline-activated persulfate oxidation, KOH or NaOH is mixed with persulfate solution to achieve the required pH.27 Since aquifers usually have a buffering capacity, a large amount of base may be needed to increase the pH of an aquifer, which increases the cost of alkaline-activated persulfate oxidation. Therefore, low-cost activators are required to render alkaline-activated persulfate more applicable in field. Basic oxygen furnace (BOF) slag is a waste material that is generated in the basic oxygen furnace steel manufacturing process. BOF slag contains 30–55% CaO, which yields hydroxyl ions that increase the pH of water rapidly, as shown in reaction (15):39,40
| | |
CaO + H2O → Ca2+ + 2OH−
| (15) |
Therefore, it may be feasible to use BOF slag to activate persulfate. The use of BOF slag to activate persulfate also allows the reuse of waste materials.
Although alkaline-activated persulfate has been used to treat various contaminants, the performance is variable. Some reports note that the removal of contaminants is enhanced by alkaline persulfate and other reports show that the use of alkaline-activated persulfate is the least efficient way to degrade pollutants.26,29,35,38 Recent studies have focused on the effectiveness of alkaline-activated persulfate for the removal of different individual compounds34,35,37 and comparisons of the degradation of individual pollutants using different persulfate activation methods.29,38 Different experimental parameters were used in other studies, such as the target pollutant, the persulfate dosage and the temperature, so it is difficult to compare the performance of each method, in terms of the removal of different pollutants by alkaline persulfate. There are also few detailed comparative studies of the removal of pollutants by alkaline persulfate that have different properties (e.g., chlorinated and petroleum hydrocarbons).26 Since 1,2-DCA and MTBE are typical chlorinated and petroleum hydrocarbons that are found in groundwater, these two compounds are the target pollutants for this study. The main objective of this study is to determine the kinetics and the mechanism for the degradation of 1,2-DCA and MTBE by alkaline-activated persulfate. Contaminants are treated using persulfate under un-activated and alkaline-activated conditions. The feasibility of using BOF slag to activate persulfate is also determined.
2. Materials and methods
2.1 Materials and chemicals
The chemicals that are used in this study are 1,2-DCA (99.9%, Merck, Germany), MTBE (99.9%, Merck, Germany), vinyl chloride (VC) (2000 ppm in methanol, Supelco, USA), tert-butyl formate (TBF) (99%, Aldrich, USA), tert-butyl alcohol (TBA) (99%+, Alfa Aesar, USA), and sodium persulfate (>99%, Riedel-de Haen, Germany). BOF slag was obtained from the China Steel Corp., Taiwan. The major constituents of BOF slag are CaO (54.4 wt%), Fe2O3 (30.6 wt%), SiO2 (9.9 wt%) and MnO (3.4 wt%). The slag was sieved before use, to ensure a more uniform particle size (10–18 mesh).
2.2 Batch kinetic experiments
Batch experiments were conducted using 60 mL serum bottles, in order to evaluate the removal of 1,2-DCA and MTBE by persulfate under un-activated and different alkaline conditions (pH 10–13). The solution used for the batch experiments was prepared by adding approximately 99 mg of 1,2-DCA or 88 mg of MTBE to 2 L DI water and 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg of persulfate was then added. 50 mL of the mixed solution was then immediately transferred to each serum bottle, using a dispenser. The initial concentrations of the contaminant and persulfate in the batch bottles were 0.5 mM and 42 mM, respectively. The pH for the batch experiments was adjusted using NaOH. BOF slag (20–100 g L−1) was used to determine whether it increases pH, which allows the alkaline-activated persulfate reaction to proceed. In a dark chamber at a constant temperature at 25 °C, batch bottles were placed on a shaker that operated at 150 rpm. All batch experiments were conducted in duplicate. Since BOF slag can contain heavy metals, such as Cr, heavy metals of interest were analyzed in the experiments where slag was added, in order to determine whether heavy metals are released from BOF slag.
000 mg of persulfate was then added. 50 mL of the mixed solution was then immediately transferred to each serum bottle, using a dispenser. The initial concentrations of the contaminant and persulfate in the batch bottles were 0.5 mM and 42 mM, respectively. The pH for the batch experiments was adjusted using NaOH. BOF slag (20–100 g L−1) was used to determine whether it increases pH, which allows the alkaline-activated persulfate reaction to proceed. In a dark chamber at a constant temperature at 25 °C, batch bottles were placed on a shaker that operated at 150 rpm. All batch experiments were conducted in duplicate. Since BOF slag can contain heavy metals, such as Cr, heavy metals of interest were analyzed in the experiments where slag was added, in order to determine whether heavy metals are released from BOF slag.
2.3 Analytical methods
Analysis of 1,2-DCA, MTBE and their degradation byproducts, including VC, TBF and TBA, was performed using a purge and trap pretreatment system (Atomx, Tekmar, USA), followed by a gas chromatograph-flame ionization detector (Agilent 7890A, USA) that was equipped with a capillary column (GsBP-624, 60 m × 0.32 mm, GS-Tek, USA). The respective operating temperatures for the injector and the detector were 180 °C and 230 °C. The oven temperature was initially maintained at 35 °C for 5 min and then increased at a rate of 11 °C min−1 to 115 °C and held at 115 °C for 3 min. The temperature was then increased at a rate of 20 °C min−1 to 220 °C and maintained at 220 °C for 1 min. The slag-activated persulfate system was analyzed for the presence of heavy metals, including Cr, Cd, Ni, Zn, Pb and Cu, using inductively coupled plasma optical emission spectrometry (ICP-OES) (Horiba, JY 2000 2, USA). The detection limits of Cr, Cd, Ni, Zn, Pb and Cu were 0.001, 0.001, 0.001, 0.002, 0.010, and 0.001 mg L−1, respectively.
3. Results and discussion
3.1 1,2-DCA degradation by alkaline-activated persulfate
Fig. 1a shows the degradation of 1,2-DCA in the alkaline-activated persulfate systems. There is significantly increased degradation of 1,2-DCA at pH values of more than 12. 98% of 1,2-DCA is removed by alkaline-activated persulfate at a pH value of 12 and 100% of 1,2-DCA is removed by alkaline-activated persulfate at a pH value of 13, but 1,2-DCA is 90% degraded by un-activated persulfate after 120 h of reaction. Although the degradation of 1,2-DCA increases as the pH value is increased, under alkaline conditions, less 1,2-DCA is removed at pH values of 10 and 11 than with un-activated persulfate. It should be noted that under alkaline controls (no addition of persulfate), 1,2-DCA at a pH of 12 and 13 produces a 4 and 11% decrease, respectively, during 120 h of reaction (data not shown in the figure). Although Block et al.26 reported that alkaline persulfate increases the degradation of 1,2-DCA, this study uses no alkaline control without persulfate. Root et al.41 noted that chloroform (CF) concentrations are reduced in both alkaline control and alkaline activated persulfate tests. The removal of CF in the control set was due to alkaline hydrolysis. The results of this study indicate that strong alkaline conditions stimulate additional degradation of 1,2-DCA, so more 1,2-DCA is removed in the alkaline persulfate systems.
 |
| | Fig. 1 (a) The degradation of 1,2-DCA and (b) the production of VC in the alkaline-activated persulfate systems. | |
Fig. 1b shows the production of VC in the alkaline-activated persulfate systems. During 1,2-DCA degradation, VC is detected in alkaline persulfate and the control runs at pH 12 and 13. VC concentrations in the control runs at pH values of 12 and 13 respectively increase to 0.023 and 0.098 mM, during 120 h of reaction. The production of VC reaches the highest concentrations of 0.006 and 0.010 mM in the alkaline persulfate systems at respective pH values of 12 and 13, after 48 h of reaction, and then gradually decrease to less than 0.001 mM because of persulfate oxidation. Jeffers et al.42 reported that VC is produced in 1,2-DCA solution at high pH values. The decrease in 1,2-DCA and the production of VC in the control sets may result from the dehydrochlorination of 1,2-DCA.42–44 Dehydrochlorination removes hydrogen and chloride ions from an alkane and results in the formation of an alkene under strong basic conditions.45,46 The transformation of 1,2-DCA to VC at high pH values is shown as below:
| |
ClH2C–CH2Cl + OH− → H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CHCl + Cl− + H2O CHCl + Cl− + H2O
| (16) |
To the best of the authors' knowledge, this is first time that the dehydrochlorination of 1,2-DCA to VC has been demonstrated in an alkaline persulfate system. Since chlorinated hydrocarbons can undergo base-mediated degradation, such as base-catalyzed hydrolysis or dehydrochlorination,42,43 it is necessary to consider the possible contribution of alkaline degradation to the removal of contaminants when alkaline persulfate is used for the treatment of chlorinated compounds.
The pseudo-first-order rate constants for 1,2-DCA degradation by un-activated and alkaline-activated persulfate are shown in Table 1. The degradation of 1,2-DCA by persulfate fits well with the pseudo-first-order kinetics (R2 > 0.96). The rate constants for the removal of 1,2-DCA at pH values of 12 and 13 are approximately 1.7 and 3.1 times greater than that for un-activated persulfate. It should be noted that although the total amount of 1,2-DCA degradation by persulfate after 120 h is not much different with or without activation, the rate of 1,2-DCA degradation is increased by alkaline activation. Since higher concentration of 1,2-DCA has higher toxicity, it is beneficial to the environment to reduce 1,2-DCA concentration rapidly during a short time. These results demonstrate that a pH value of greater than 12 is required to increase 1,2-DCA degradation in alkaline persulfate systems. Since sulfate radicals and hydroxyl radicals are generated in alkaline-activated persulfate systems,29 better performance may result from the synergistic contribution of oxidation and dehydrochlorination. The VC that is produced by the dehydrochlorination of 1,2-DCA is subsequently removed by the oxidative radicals.
Table 1 The results for the degradation of contaminants by un-activated and alkaline-activated persulfate
| Contaminant (mM) |
Persulfate (mM) |
pH |
k × 103 (h−1) |
R2 |
Final pH |
Final ORP (mV) |
Corrected ORP for pH 7 |
| 0.5 (1,2-DCA) |
42 |
4 (un-activated) |
13.6 |
0.993 |
2.4 |
760 |
488 |
| 10 |
5.2 |
0.962 |
3.2 |
706 |
481 |
| 11 |
9.0 |
0.992 |
11.2 |
356 |
604 |
| 12 |
23.7 |
0.976 |
12.5 |
389 |
714 |
| 13 |
42.1 |
0.994 |
13.2 |
449 |
815 |
| 0.5 (MTBE) |
4 (un-activated) |
12.6 |
0.991 |
2.7 |
652 |
398 |
| 10 |
4.2 |
0.990 |
3.3 |
625 |
406 |
| 11 |
5.2 |
0.997 |
11.6 |
278 |
550 |
| 12 |
7.4 |
0.992 |
12.7 |
347 |
684 |
| 13 |
6.6 |
0.997 |
13.3 |
463 |
835 |
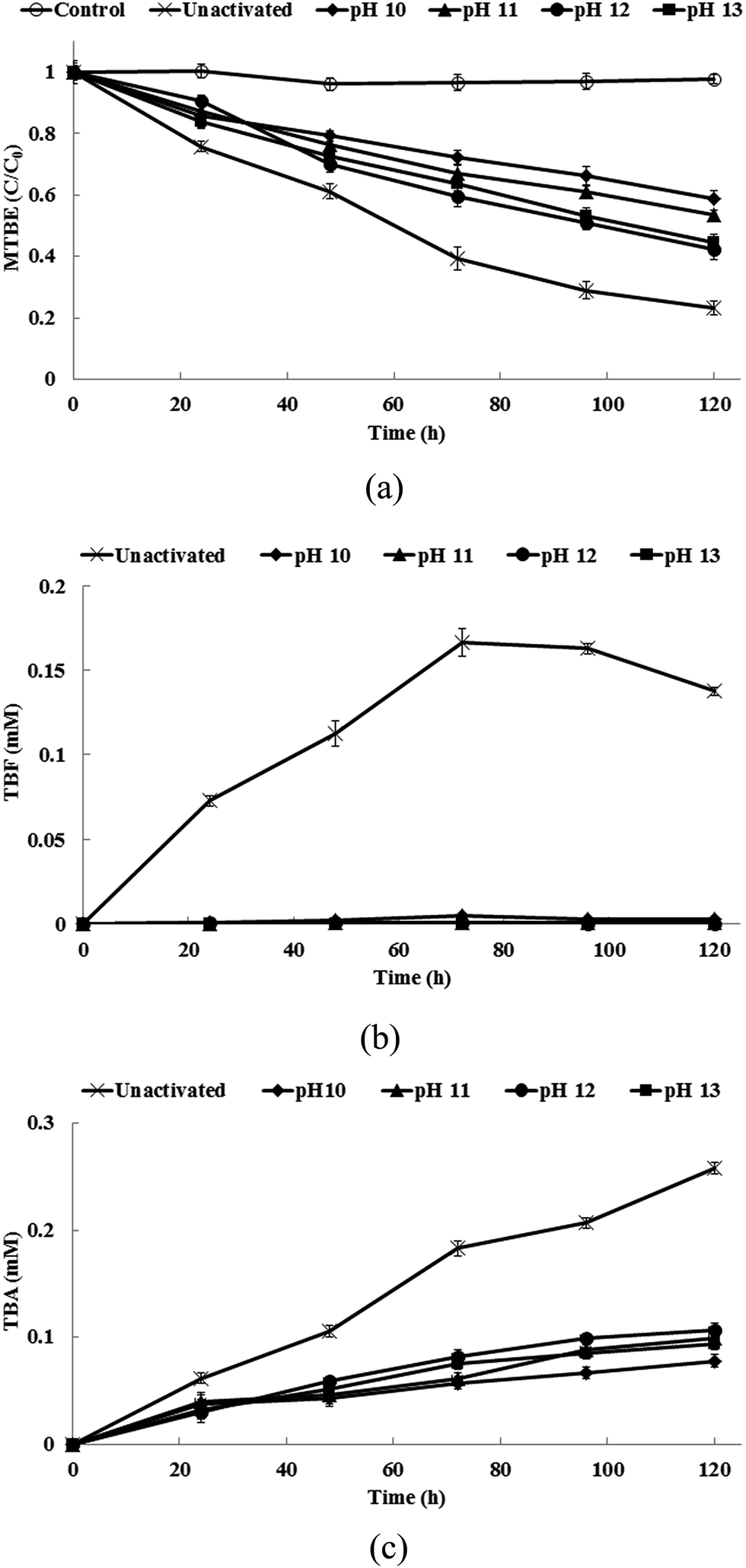
3.2 MTBE degradation by alkaline-activated persulfate
Fig. 2a shows the degradation of MTBE in the alkaline-activated persulfate systems. MTBE degradation was retarded under all alkaline conditions. 77% of MTBE is removed in un-activated persulfate, but only 41–58% of MTBE is degraded at pH values between 10 and 13. The degradation of MTBE is also greater at pH values of 12 and 13 than at pH values of 10 and 11. These findings are in agreement with those of a previous study that was conducted by Deng et al.29 That study reported that the removal of MTBE under alkaline conditions is much slower than by persulfate alone at 25 °C. Huang et al.36 also noted that the rate at which MTBE is degraded by thermally activated persulfate (40 °C) decreases as the pH value increases (pH 2.5–12). No MTBE degradation is observed for the alkaline control sets (data not shown in the figure). Therefore, only persulfate oxidation is responsible for the removal of MTBE by alkaline persulfate. The pseudo-first-order rate constants for the degradation of MTBE by un-activated and alkaline-activated persulfate are shown in Table 1. The degradation of MTBE by persulfate follows the pseudo-first-order kinetics (R2 > 0.99). Un-activated persulfate gives the greatest removal rate for MTBE. This rate is approximately 1.7 to 3.0 times greater than the rate for alkaline persulfate.
 |
| | Fig. 2 (a) The degradation of MTBE, (b) the production of TBF and (c) the production of TBA in the alkaline-activated persulfate systems. | |
During the oxidation of MTBE, the byproducts, TBF and TBA, are produced. Fig. 2b and c respectively show the production of TBF and TBA in the alkaline-activated persulfate systems. Notably, the final concentrations of TBF (0–0.003 mM) in the alkaline systems are much lower than that in the un-activated system (0.137 mM). TBF is a major byproduct of the oxidation of MTBE. The TBF that is produced subsequently undergoes hydrolysis to yield TBA.36 Church et al.47 noted that the respective rate constants for the hydrolysis of TBF to TBA under basic, neutral and acid and conditions are 1.7 ± 0.3 M−1 s−1, (1.0 ± 0.2) × 10−6 s−1 and (2.7 ± 0.5) × 10−3 M−1 s−1. The rate constant for the basic hydrolysis of TBF is 500 times greater than that for the acid hydrolysis of TBF. Therefore, the TBF that is produced in the alkaline persulfate systems is rapidly converted to TBA, so less TBF accumulates. The concentrations of TBA in the alkaline persulfate systems are also significant lower than that in the un-activated system, which is consistent with the results of the study by Deng et al.,29 which showed that alkaline persulfate degrades TBA more effectively than persulfate alone at 25 °C. It has been reported that TBF hydrolysis generates the same stoichiometric amount of TBA.47 Therefore, the final concentrations of TBF and TBA can be added and expressed as “mM as TBA”. Table 2 shows the molar ratio of TBF and TBA accumulation/MTBE removal in the un-activated and alkaline-activated persulfate systems after 120 h of reaction. At the end of the experiments, the molar ratios of TBF and TBA accumulation/MTBE removal in the alkaline-activated persulfate systems range from 0.30 to 0.40 and the molar ratio for TBF and TBA accumulation/MTBE removal is 0.98 in the un-activated persulfate system. These results show that alkaline-activated persulfate reduces the production of byproducts during MTBE degradation. Since TBA is a known toxin and an animal carcinogen and it is the only degradation byproduct that has the potential to persist in the environment,47,48 the use of alkaline persulfate is beneficial because it reduces the accumulation of this harmful byproduct.
Table 2 The molar ratio of TBF and TBA production to MTBE removal for un-activated and alkaline-activated persulfate systems after 120 h of reaction
| pH |
MTBE removal (mM) |
Final TBF (mM) |
Final TBA (mM) |
TBF + TBA (mM as TBA) |
TBF/TBA production (mM)/MTBE removal (mM) |
| 4 (un-activated) |
0.41 |
0.14 |
0.26 |
0.40 |
0.98 |
| 10 |
0.22 |
0.003 |
0.083 |
0.086 |
0.39 |
| 11 |
0.25 |
0 |
0.10 |
0.10 |
0.40 |
| 12 |
0.31 |
0 |
0.11 |
0.11 |
0.35 |
| 13 |
0.30 |
0 |
0.09 |
0.09 |
0.30 |
3.3 The effect of pH and ORP on the removal of contaminants
Table 1 shows that the pH values for the sets with pH values of 11, 12 and 13 are maintained at the required pH during the experiments, in order to evaluate 1,2-DCA and MTBE oxidation. However, the pH values for the sets for pH 10 are maintained at pH 10 for 48 h of reaction and then gradually decrease to around 3.2. The ORP values, measured for the sets for pH 11, 12 and 13 are stable during the experiments and the final ORP for these sets is much lower than that for un-activated persulfate. In general, H+ concentration affects the measured redox potential according to eqn (17):34| | |
Emeasured = K − 0.059pH
| (17) |
where K is a constant and the unit of K and E is volts. Eqn (17) shows that ORP decreases as the pH increases. Even if the oxidative radicals are generated by alkaline-activated persulfate, the ORP may decrease because of the effect of a high pH. Therefore, it is necessary to correct the ORP that is measured at different pH values to the value for the same pH, in order to determine the real effect of alkaline persulfate on the system ORP. Using eqn (17), the ORP measured at different pH values is corrected to a reference pH, as shown eqn (18):| | |
Ecorrected = Emeasured − 0.059(pHreference − pHmeasured)
| (18) |
In this study, the ORP levels at different pH values are corrected for a pH of 7. As shown in Table 1, the corrected ORP values for the sets for pH 11, 12 and 13 are much greater than that for un-activated persulfate when the effect of pH on the ORP measurement is eliminated. Liang et al.28,34 reported the presence of hydroxyl radicals (predominant) and sulfate radicals under strong alkaline conditions (pH > 11). Therefore, the production of the oxidative radicals by alkaline persulfate (reactions (4), (9), (13) and (14)) contributes to the oxidative power of the system. Otherwise, the measured ORP in the high pH systems would be even lower than the measured ORP that is shown in Table 1.
Under alkaline conditions (pH 11 to 13), both the measured and the corrected ORP increase as the pH value increases. An increase in pH also results in greater removal of 1,2-DCA and MTBE, especially at pH values of 12 and 13. This demonstrates that higher pH values result in stronger persulfate activation. The removal of 1,2-DCA and MTBE is least efficient at pH 10. The corrected and measured ORP values for the sets for pH 10 are similar to that for un-activated persulfate. Therefore, the high ORP for the sets for pH 10 mainly results from the low pH and the un-activated persulfate. Although the pH is initially maintained at around 10, alkaline activation of persulfate is not as strong as that for the sets where the pH is greater than 11, so insufficient radicals are produced for the oxidation of 1,2-DCA and MTBE. Since the pH gradually decreases, there is a moderate increase in the ORP, so 1,2-DCA and MTBE do not stay in a high ORP solution for the duration of the experiments. There is also no degradation of 1,2-DCA or MTBE at a pH value of 10, as shown by the results for the alkaline controls. These may be the reasons that less contaminants are removed at a pH value of 10. Note that in reality, the pH has a dominant effect on ORP value in the persulfate solution. If no other mechanisms contribute to the degradation of contaminants, the efficiency with which the contaminant is treated decreases at high pH because ORP values are low. This may explain why less contaminant is removed in many other reports that use alkaline-activated persulfate for the treatment.27
Some studies note that the removal of contaminants is increased by alkaline persulfate because hydroxyl radicals are produced,34,35 but other studies report that sulfate radicals are scavenged by hydroxyl ions and the hydroxyl radicals that are produced are rapidly consumed by sulfate ions, hydroxyl ions and other reactants in the alkaline persulfate systems, so the contaminants experience less degradation.32,36,37,49 However, no direct evidence that affects the degradation of pollutants is provided by previous reports and relatively few studies concern the relationship between contaminant removal, pH and ORP.34 The results for the ORP measurement may explain the poor performance of alkaline persulfate in the removal of some contaminants.
3.4 1,2-DCA and MTBE degradation by BOF slag-activated persulfate
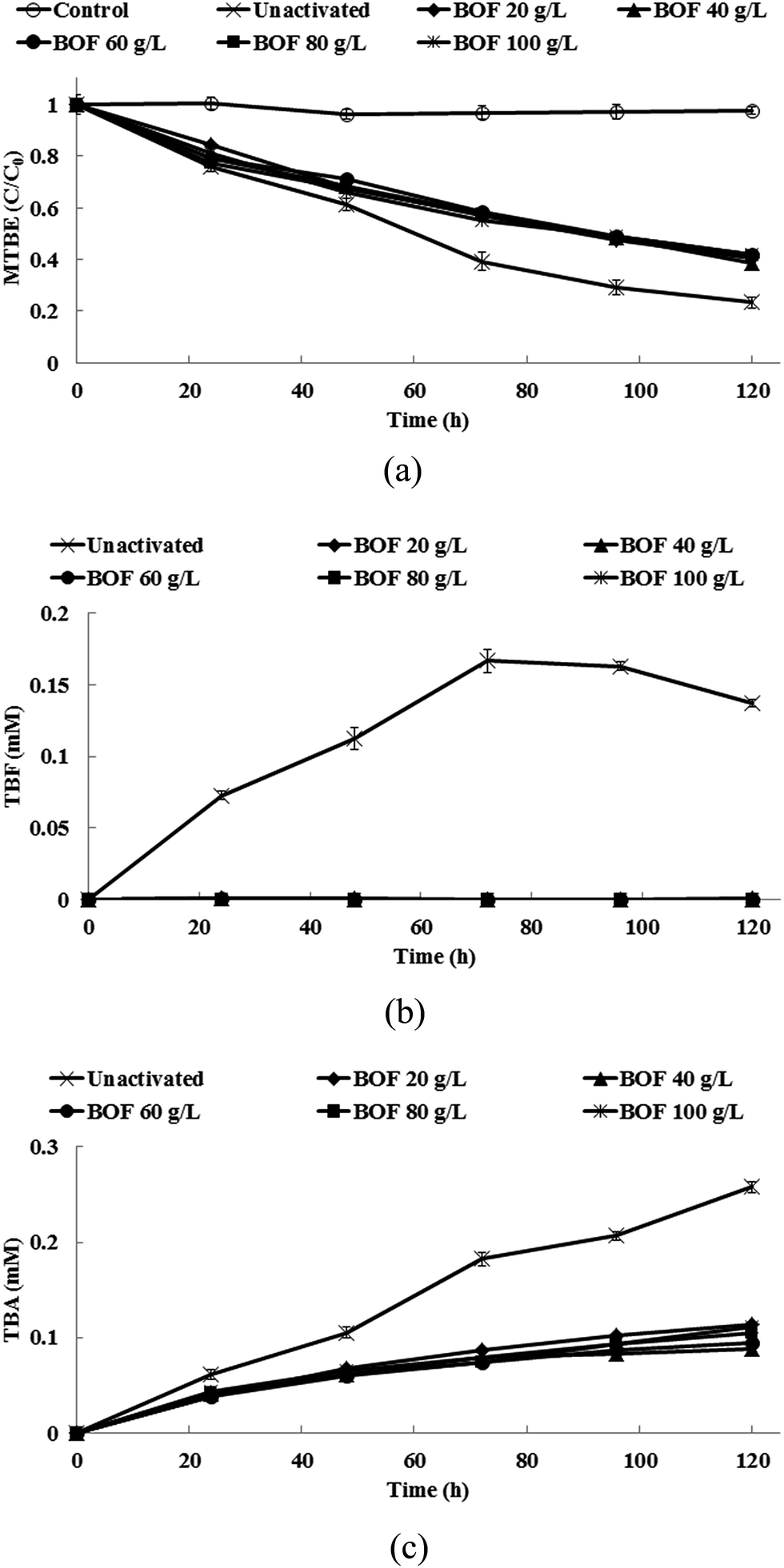
Fig. 3a shows the degradation of 1,2-DCA by persulfate when different dosages of BOF slag are added. When BOF slag is added, the pH in the system rapidly increases and then remains between 12.3 and 12.6 during the experiments. 100% of 1,2-DCA is removed for different dosages of BOF. A small amount of VC is also produced during the degradation of 1,2-DCA and this is then completely removed at the end of the experiments (Fig. 3b). The addition of 20 g L−1 slag or more increases the pH and increases the degradation of 1,2-DCA. Table 3 shows the pseudo-first-order rate constants for the degradation of 1,2-DCA and MTBE by BOF slag-activated persulfate. The rate constants for the removal of 1,2-DCA by BOF slag-activated persulfate are approximately 2.4–2.7 times greater than that for un-activated persulfate. Fig. 4a shows the degradation of MTBE by persulfate when different dosages of BOF slag are added. Less MTBE is degraded at high pH values. The removal rate for MTBE by un-activated persulfate is 1.7 to 1.8 times greater than that for persulfate that is activated by BOF slag (Table 3). As shown in Fig. 4b and c, the accumulation of TBF and TBA is mitigated by the addition of BOF slag because the pH is high. The results of the heavy metal analysis show that no detectable amounts of heavy metals are found during treatment with persulfate that is activated by BOF slag. Therefore, it is safe to use BOF slag as an activator for persulfate.
 |
| | Fig. 3 (a) The degradation of 1,2-DCA and (b) the production of VC in the persulfate systems that are activated by slag. | |
Table 3 The rate constants for the degradation of 1,2-DCA and MTBE by persulfate that is activated by BOF slag
| Contaminant (mM) |
Persulfate (mM) |
BOF slag (g L−1) |
Final pH |
k × 103 (h−1) |
R2 |
| 0.5 (1,2-DCA) |
42 |
0 (un-activated) |
2.4 |
13.6 |
0.993 |
| 20 |
12.3 |
32.5 |
0.974 |
| 40 |
12.6 |
34.7 |
0.954 |
| 60 |
12.6 |
36.7 |
0.954 |
| 80 |
12.6 |
33.8 |
0.970 |
| 100 |
12.6 |
34.8 |
0.968 |
| 0.5 (MTBE) |
0 (un-activated) |
2.6 |
12.6 |
0.991 |
| 20 |
12.4 |
7.6 |
0.998 |
| 40 |
12.6 |
7.6 |
0.995 |
| 60 |
12.6 |
7.1 |
0.994 |
| 80 |
12.7 |
7.1 |
0.991 |
| 100 |
12.7 |
7.1 |
0.993 |
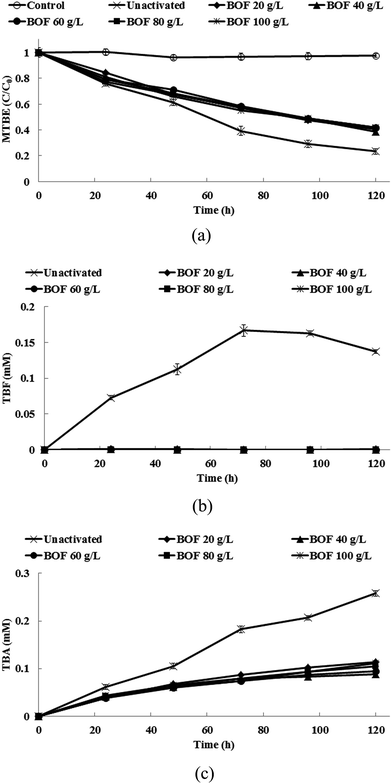 |
| | Fig. 4 (a) The degradation of MTBE, (b) the production of TBF and (c) the production of TBA in the persulfate systems that are activated by slag. | |
The removal of 1,2-DCA and MTBE and the production of the byproducts by persulfate that is activated by BOF slag are quite similar to the results alkaline-activate persulfate using NaOH as the activator. This study's findings demonstrate that BOF slag is a good alternative for the alkaline-activated persulfate reaction. Although BOF slag contains a significant amount of iron oxide, iron oxide is not involved with the activation of persulfate because iron ions are only released with difficulty at high pH values. BOF slag has been successfully used to catalyze hydrogen peroxide in Fenton-like systems, under acid conditions. The homogeneous catalytic reaction of hydrogen peroxide by the Fe2+ that is leached from BOF slag plays a dominant role in increasing the removal of contaminants and the heterogeneous reaction of hydrogen peroxide with the iron that is contained in BOF slag plays a minor role.50 Liang et al.49 reported that the use of BOF slag (1 g L−1) results in an increase in the pH of the solution to 11, which is unfavorable to a Fenton-like reaction because iron ions are not produced at such a high pH value. Tsai et al.51 noted that less TCE is removed as the pH value increases in a H2O2/BOF slag system because there is less dissociation of iron minerals from BOF slag. Teel et al.52 reported that iron oxide hematite (Fe2O3) does not activate persulfate to generate oxidative radicals. Therefore, hydroxyl ions (eqn (15)) that are released from BOF slag are the main contributors to the activation of persulfate, rather than the dissolved iron ions or the iron oxide on the surface of the BOF slag.
4. Conclusions
In this study, 1,2-DCA and MTBE is removed using alkaline-activated persulfate, with NaOH and BOF slag as activators. The results show that 1,2-DCA removal is increased when alkaline-activated persulfate is used at pH values greater than 12. VC is generated due to the dehydrochlorination of 1,2-DCA in the alkaline-activated persulfate systems. Although MTBE degradation decreases, the accumulation of TBF and TBA is significantly reduced when alkaline-activated persulfate is used. Alkaline-activated persulfate degrades 1,2-DCA via the mechanisms of oxidation and dehydrochlorination and oxidation alone contributes to the removal of MTBE. Low-cost BOF slag (>20 g L−1) increases the pH of the solution and drives the alkaline-activated persulfate reaction. No detectable amounts of heavy metals are found during the experiments, which demonstrates that it is safe to use BOF slag for the alkaline activation of persulfate. The removal of 1,2-DCA and MTBE in un-activated systems and those that are activated by NaOH and BOF obeys the pseudo-first-order kinetics. Although a high pH can cause a decrease in ORP, which is not favorable to the oxidation of contaminants, sulfate and hydroxyl radicals that are generated by alkaline activation of persulfate increase the ORP in the alkaline solution and still play an important role in the removal of contaminants. The use of alkaline-activated persulfate also reduces the potential for the release of heavy metals from aquifer sediments during site remediation. Since alkaline-activated persulfate decreases the removal rate for MTBE, it is necessary to evaluate the performance in terms of the removal of contaminants before alkaline-activated persulfate is used for other target compounds. This study demonstrates a novel alkaline-activated system that uses BOF slag as the activator to treat groundwater contaminants. The results of this study will allow the design of an environmentally friendly and cost-effective alkaline persulfate system for groundwater remediation.
Acknowledgements
This research was funded by the Ministry of Science and Technology, Taiwan (103-2221-E-260-001-MY3; 104-2622-E-260-003-CC3). Additional thanks are extended to the personnel and graduate students at National Chi Nan University, Taiwan, for their assistance throughout this project.
References
- U. S. EPA, Cleaning Up the Nation's Waste Sites Markets and Technology, EPA 542-R-04–015, 2004 Search PubMed.
- U. S. EPA, How to Evaluate Alternative Cleanup Technologies for Underground Storage Tank Sites: A Guide for Corrective Action Plan Reviewers, EPA 510-R-04–002, 2004 Search PubMed.
- K. F. Chen, C. M. Kao, J. Y. Wang, T. Y. Chen and C. C. Chien, J. Hazard. Mater., 2005, 125, 10–16 CrossRef CAS PubMed.
- S. Y. Wang, Y. C. Kuo, Y. Z. Huang, C. W. Huang and C. M. Kao, Environ. Pollut., 2015, 203, 97–106 CrossRef CAS PubMed.
- U. S. EPA, Integrated Risk Information System (IRIS) Chemical Assessment Summary: 1,2-Dichloroethane Carcinogenicity Assessment (CASRN 107-06-2), 1987 Search PubMed.
- I. A. R. C. (International Agency for Research on Cancer), IARC Monogr. Eval. Carcinog. Risks Hum., 1999, 71, 501–529 Search PubMed.
- U. S. EPA, EPA Protocol for the Second Six-Year Review (Updated), EPA 815-B-09–002, 2009 Search PubMed.
- K. F. Chen, C. M. Kao, L. C. Wu and S. H. Liang, Water Environ. Res., 2009, 81, 687–694 CrossRef CAS PubMed.
- U. S. EPA, Drinking Water Advisory: Consumer Acceptability Advice and Health Effects Analysis on Methyl Tertiary-butyl Ether (MTBE), EPA-822-F-97–009, D.C., 1997 Search PubMed.
- S. G. Huling and B. E. Pivetz, Engineering Issue: In-Situ Chemical Oxidation, EPA-600-R-06–072, U. S. Environmental Protection Agency, 2006 Search PubMed.
- B. A. Kocamemi and F. Cecen, Int. Biodeterior. Biodegrad., 2009, 63, 717–726 CrossRef.
- K. F. Chen, Y. C. Chang and S. C. Huang, Desalin. Water Treat., 2012, 48, 278–284 CrossRef CAS.
- S. Ko, M. Crimi, B. K. Marvin, V. Holmes and S. G. Huling, J. Environ. Manage., 2012, 108, 42–48 CrossRef CAS PubMed.
- I. T. R. C. (Interstate Technology & Regulatory Council), Technical and Regulatory Guidance for In Situ Chemical Oxidation, 2nd edn, 2005 Search PubMed.
- R. L. Johnson, P. G. Tratnyek and R. O. Johnson, Environ. Sci. Technol., 2008, 42, 9350–9356 CrossRef CAS PubMed.
- J. Costanza, G. Otano, J. Callaghan and K. D. Pennell, Environ. Sci. Technol., 2010, 44, 9445–9450 CrossRef CAS PubMed.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1213–1223 CrossRef CAS PubMed.
- C. Liang, C. J. Bruell, M. C. Marley and K. L. Sperry, Chemosphere, 2004, 55, 1225–1233 CrossRef CAS PubMed.
- C. Liang, C. F. Huang and Y. J. Chen, Water Res., 2008, 42, 4091–4100 CrossRef CAS PubMed.
- S. Y. Oh, H. W. Kim, J. M. Park, H. S. Park and C. Yoon, J. Hazard. Mater., 2009, 168, 346–351 CrossRef CAS PubMed.
- I. Hussain, Y. Zhang, S. Huang and X. Du, Chem. Eng. J., 2012, 203, 269–276 CrossRef CAS.
- Y. T. Lin, C. Liang and J. H. Chen, Chemosphere, 2011, 82, 1168–1172 CrossRef CAS PubMed.
- Y. Q. Gao, N. Y. Gao, Y. Deng, Y. Q. Yang and Y. Ma, Chem. Eng. J., 2012, 195–196, 248–253 CrossRef CAS.
- S. Yang, X. Yang, X. Shao, R. Niu and L. Wang, J. Hazard. Mater., 2011, 186, 659–666 CrossRef CAS PubMed.
- P. A. Block, R. A. Brown and D. Robinson, Novel activation technologies for sodium persulfate in situ chemical oxidation, Proceedings of the 4th International Conference on the Remediation of Chlorinated and Recalcitrant Compounds, 2004 Search PubMed.
- A. Tsitonaki, B. Petri, M. Crimi, H. Mosbaek, R. L. Siegrist and P. L. Bjerg, Crit. Rev. Environ. Sci. Technol., 2010, 40, 55–91 CrossRef CAS.
- H. Y. Liang, Y. Q. Zhang, S. B. Huang and I. Hussain, Chem. Eng. J., 2013, 218, 384–391 CrossRef CAS.
- D. Deng, L. Peng, M. Guan and Y. Kang, J. Hazard. Mater., 2014, 264, 521–528 CrossRef CAS PubMed.
- J. F. Corbin III, Mechanisms of Base, Mineral, and Soil Activation of Persulfate for Groundwater Treatment, A dissertation submitted in partial fulfillment of the requirements for the degree of Doctor of Philosophy, Washington State University, 2008.
- R. J. Watts, Enhanced Reactant-contaminant Contact through the Use of Persulfate In Situ Chemical Oxidation (ISCO), SERDP Project ER-1489, 2011 Search PubMed.
- G. D. Fang, D. D. Dionysiou, D. M. Zhou, Y. Wang, X. D. Zhu, J. X. Fan, L. Cang and Y. J. Wang, Chemosphere, 2013, 90, 1573–1580 CrossRef CAS PubMed.
- O. S. Furman, A. L. Teel and R. J. Watts, Environ. Sci. Technol., 2010, 44, 6423–6428 CrossRef CAS PubMed.
- C. Liang, C. P. Liang and C. C. Chen, J. Contam. Hydrol., 2009, 106, 173–182 CrossRef CAS PubMed.
- C. Liang and H. W. Su, Ind. Eng. Chem. Res., 2009, 48, 5558–5562 CrossRef CAS.
- K. C. Huang, R. A. Couttenye and G. E. Hoag, Chemosphere, 2003, 49, 413–420 CrossRef.
- S. G. Huling, S. Ko, S. Park and E. Kan, J. Hazard. Mater., 2011, 192, 1484–1490 CrossRef CAS PubMed.
- D. Zhao, X. Liao, X. Yan, S. G. Huling, T. Chai and H. Tao, J. Hazard. Mater., 2013, 254–255, 228–235 CrossRef CAS PubMed.
- L. de Windt, P. Chaurand and J. Rose, Waste Manage., 2011, 31, 225–235 CrossRef CAS PubMed.
- C. Shi, J. Mater. Civ. Eng., 2004, 16, 230–236 CrossRef CAS.
- D. K. Root, E. M. Lay, P. A. Block and W. G. Cutler, Investigation of chlorinated methanes treatability using activated sodium persulfate, Proceedings of the First International Conference on Environmental Science and Technology, 2005 Search PubMed.
- P. M. Jeffers, L. M. Ward, L. M. Woytowltch and N. L Wolfe, Environ. Sci. Technol., 1989, 23, 965–969 CrossRef CAS.
- J. W. Washington, Ground Water, 1995, 33, 415–424 CrossRef CAS.
- P. M. Jeffers and N. L. Wolfe, Environ. Toxicol. Chem., 1996, 15, 1066–1070 CAS.
- K. A. Marshall, Chlorocarbons and Chlorohydrocarbons, Survey, in Kirk-Othmer Encyclopedia of Chemical Technology, John Wiley & Sons, Inc., New York, NY, 2003, vol. 6, pp. 226–253 Search PubMed.
- U. S. G. S. (US Geological Survey), Description, Properties, and Degradation of Selected Volatile Organic Compounds Detected in Ground Water— A Review of Selected Literature, Open-File Report 2006-1338, 2006 Search PubMed.
- C. D. Church, J. F. Pankow and P. C. Tratnyek, Environ. Toxicol. Chem., 1999, 18, 2789–2796 CrossRef CAS.
- W. Q. Zhuang, J. H. Tay, S. Yi and S. T. Tay, J. Biotechnol., 2005, 118, 45–53 CrossRef CAS PubMed.
- J. Liang, S. Komarov, N. Hayashi and E. Kasai, Ultrason. Sonochem., 2007, 14, 201–207 CrossRef CAS PubMed.
- C. S. Chiou, C. F. Chang, C. T. Chang, J. L. Shie and Y. H. Chen, Chemosphere, 2006, 62, 788–795 CrossRef CAS PubMed.
- T. T. Tsai, C. M. Kao and J. Y. Wang, Chemosphere, 2011, 83, 687–692 CrossRef CAS PubMed.
- A. L. Teel, M. Ahmad and R. J. Watts, J. Hazard. Mater., 2011, 196, 153–159 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 *,
Yu-Chen Chang and
Kuan-Yu Liu
*,
Yu-Chen Chang and
Kuan-Yu Liu