DOI:
10.1039/C6RA15952D
(Paper)
RSC Adv., 2016,
6, 96250-96262
Novel approach of adaptive laboratory evolution: triggers defense molecules in Streptomyces sp. against targeted pathogen
Received
20th June 2016
, Accepted 27th September 2016
First published on 27th September 2016
Abstract
The adaptation and evolution of microorganisms under selective pressure is the major cause of the development of antibiotic resistance. However, our present study presents adaptive laboratory evolution as an efficient tool to mine for and develop targeted bioactive molecules. Cryptococcal meningitis is an emerging neurological disease with limited therapeutic options. In the present study, a new marine anticryptococcal strain of Streptomyces variabilis AFP2 was co-cultured with C. neoformans with periodic transfer for 30 days. AFP2 displayed improved anticryptococcal activity post co-culture versus the parental type. The metabolomes of the parental and evolved strains were analyzed by HPLC-UV and GC-MS. The changes in phenotype and chemotype between the parental and the evolved strain were analyzed to determine the evolution of new traits. About 21 new metabolites alien to the parent strain were seen to be produced by the evolved strain. Among these, a few of the induced molecules such as DL-alanyl-L-leucine, dihydro-3,3-dimethyl-2(3H)-furanone, enanthamide, 1,3,5-cycloheptatriene and 1-aziridineethanol were not reported to have antifungal activity. In addition, these molecules were not reported in S. variabilis. Evolutionary fitness analysis revealed a 64-fold or 98% reduction in the growth of C. neoformans for the evolved strain S3, with an overexpression of compounds of ∼52%. Hence, the present study confirms that an integrated approach using adaptive laboratory techniques and co-culturing techniques is useful to discover potential molecules active against targeted pathogens.
Introduction
Invasive fungal infections have emerged as a great challenge in medicine. Their incidence and mortality rates have dramatically increased in the last few decades.1 Although patients with weak immunity are highly susceptible and prone to these infections, reports of healthy individuals being affected are also on the rise. The most common causes of invasive fungal infections in healthy individuals are surgery and long-term antibiotic treatment.2–4 Cryptococcus neoformans is an emerging opportunistic fungal pathogen that causes meningitis, a neurological infection, in immunocompromised patients and is particularly predominant in patients with HIV.5,6 C. neoformans has unique virulence properties such as capsule production and melanization during infection. Although the availability of antiretroviral therapy has helped patients to become less prone to cryptococcal infection, it is still a major problem under resource-limited conditions where HIV is still prevalent.7 Recently, the Centers for Disease Control estimated that there are approximately one million new cases of cryptococcal meningitis each year, which result in the death of 625![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people worldwide.8,9 Therapeutic options for the management of cryptococcal infections are limited. The current therapy consists of a cocktail of amphotericin B and flucytosine (5-FC), but 5-FC remains largely unavailable in Asia and Africa where cryptococcal meningitis is common.10 However, fluconazole intake for a prolonged period of time can lead to other yeast infections such as fluconazole-resistant candidiasis, whereas in the case of highly efficient lipid-based amphotericin B its cost and inability to pass through the blood–brain barrier are major setbacks. Thus, the need for research into novel, effective and safe drugs to treat cryptococcal meningitis has become a relevant question in the scientific community.11,12
000 people worldwide.8,9 Therapeutic options for the management of cryptococcal infections are limited. The current therapy consists of a cocktail of amphotericin B and flucytosine (5-FC), but 5-FC remains largely unavailable in Asia and Africa where cryptococcal meningitis is common.10 However, fluconazole intake for a prolonged period of time can lead to other yeast infections such as fluconazole-resistant candidiasis, whereas in the case of highly efficient lipid-based amphotericin B its cost and inability to pass through the blood–brain barrier are major setbacks. Thus, the need for research into novel, effective and safe drugs to treat cryptococcal meningitis has become a relevant question in the scientific community.11,12
The majority of antimicrobial agents in clinical use are natural products obtained from microorganisms.13 The ocean constitutes one source for developing antimicrobial agents owing primarily to its rich biodiversity. Marine organisms, especially plants and invertebrates, have received a lot of attention owing to their ability to produce antimicrobial agents. Along with marine plants and animals, marine microbes, which are found to be distributed in every niche of the ocean ecosystem, form a poorly investigated source of antimicrobial agents.14 Although new antimicrobial agents from Actinomycetes are being identified, they are nevertheless mostly related compounds or derivatives. To search for more diversified antibiotics, we have co-cultivated Streptomyces sp. with a pathogen, thereby helping to trigger defense and the related production of metabolites. It has been shown that co-cultivation is an efficient method for identifying and employing new secondary metabolites that are not detected in axenic cultures of the producing strain.15 For example, during co-cultivation fungally derived enniatin derivatives displayed an increase in antagonistic alterations against MRSA.16 Co-cultivation has been identified as a powerful emerging tool for enhancing chemical diversity in microbes. Although this technique has been found to be promising in biomedical research, it is still in its infancy.
There are various methods of elicitation that are being used by various research groups to induce secondary metabolites that were not actually expressed in monocultures such as elicitation by microbial co-cultivation,17–19 microbial lysates,20 and cellular components,21,22 chemical elicitation,23 and molecular elicitation.24 Adaptive laboratory evolution (ALE) has also been reported as a method of acceleration to induce the production of secondary metabolites.25–27
ALE is an important scientific approach in the analysis of evolutionary phenomena.28 ALE is a well-known method for generating new improved phenotypes of industrial importance. In addition, ALE is used as a laboratory technique to investigate ethanol tolerance, osmotic stress, and antibiotic resistance, and to increase the production of antibiotics.26,28–31
In the present study, we developed a novel approach to trigger and improve the activity of bioactive molecules by a combined approach using ALE and co-culture. A new strain of Streptomyces variabilis AFP2 displaying anticryptococcal activity was co-cultivated with C. neoformans. The technique was carried out for a period of 30 days, which comprised 3 serial passages of S. variabilis AFP2. The metabolomes of the wild-type and evolved strains were compared by HPLC-UV and GC-MS methods.
Methodology
Isolation of marine Actinomycetes spp.
Samples of marine sediment were collected from the sea at Rameswaram, Tamil Nadu, India (latitude: 9.145105 to 9.06412677; longitude: 79.453812 to 79.31699753) at different depths ranging from the surface to 250 cm. The sediment samples were mixed together and preserved at 4 °C until further processing. A simple pretreatment method was employed to promote the isolation of Actinomycetes spp. The soil samples were incubated at 60 °C for 40 min and resuspended in 0.9% saline water. The resuspended mixture was diluted with 100 mL saline containing 1.5% (v/v) phenol and shaken for 30 min at 28 °C.32 The pretreated samples were plated on various media such as Zobell marine agar,33 Actinomycetes isolation agar,34 starch agar,35 yeast extract–malt extract agar21 and starch–casein–nitrate agar.35 The media were supplemented with gentamycin (1 μg mL−1) and fluconazole (50 μg mL−1) after sterilization to inhibit the growth of bacteria and fungi, respectively.36,37 The plates were incubated for 21 days at 37 °C. The isolates obtained from each medium were stored in 15% glycerol at −80 °C.
Antagonism assay against C. neoformans
Test strain. The test strain C. neoformans 14116 was purchased from the Microbial Culture Collection Centre, Chandigarh. The strain was maintained in PDA slants at 4 °C and 15% glycerol stocks at −80 °C.
Antifungal assay. The anticryptococcal activity of 60 marine Actinomycetes isolates was assayed to select a potential strain according to Ramakrishnan et al. Spore suspensions of individual isolates were spot-inoculated (10 μL per spot) on Mueller–Hinton agar plates and incubated at 30 °C for 3 days. The cells were then killed with chloroform vapor and were subsequently overlaid with agar with 15 mL of a medium containing 1% (w/v) agar, 0.5% (w/v) peptone, and 0.5% (w/v) yeast extract and swabbed with 100 μL of a test isolate of C. neoformans on the agar surface. The resulting clear zone of inhibition (ZOI) was measured after incubation for 2 days. The experiment was repeated three times. The mean diameter of the ZOI and the standard deviations were calculated. The AFP2 strain, which exhibited the maximum antagonistic activity against C. neoformans, was selected for taxonomical investigation.38,39
Taxonomical investigation of AFP2. Genomic DNA from AFP2 was isolated using the procedure described by Kimura.40 Gene fragments were amplified using a PCR kit (GENEI Pvt. Ltd, India) and the 16S rRNA gene was amplified using an Eppendorf Mastercycler pro thermal cycler with the following procedure: initial denaturation at 95 °C for 4 min, 30 amplification cycles of 95 °C for 1 min, an annealing temperature of 50 °C for 60 s, and 72 °C for 1 min, and a final extension step at 72 °C for 4 min. The PCR product was electrophoresed and purified from 1.5% agarose gel using a QIAquick PCR purification kit (QIAGEN) and sequenced using the 8F and U1492R primers.41,42 Sequencing was performed at Chromous Biotech, Bangalore, India using an ABI 3100 sequencer (Applied Biosystems). The sequence was edited using FinchTV (Geospiza Inc.) and BioEdit (Ibis Biosciences, Abbott Labs). A sequence similarity search was conducted using the 16S rRNA gene and a taxid-specific BLAST tool.39
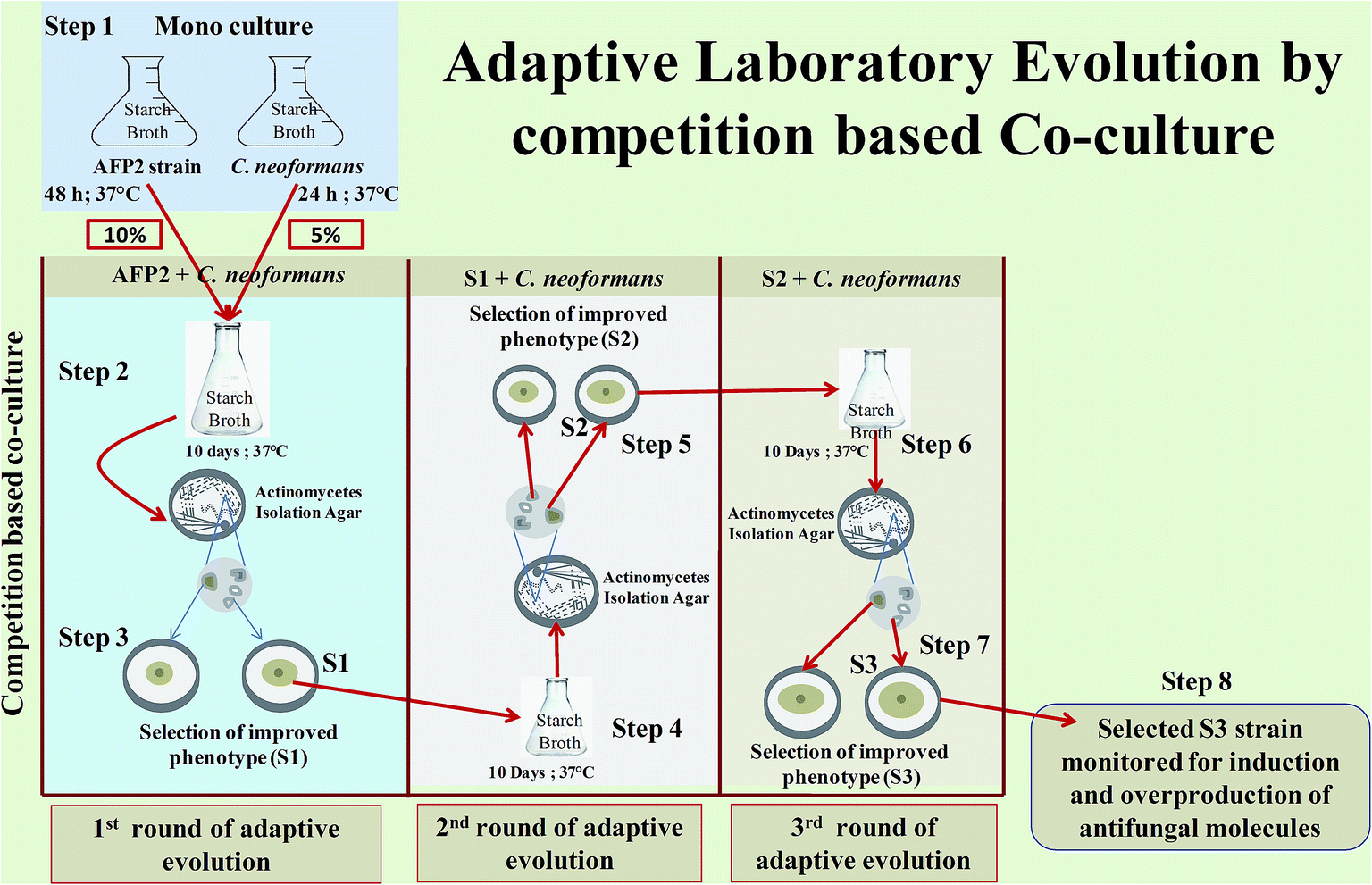
Adaptive laboratory evolution by competition-based co-culture
Adaptive laboratory evolution by competition-based co-culture was begun by growing AFP2 and C. neoformans separately in 50 mL starch broth at 37 °C for 48 h and 24 h, respectively, in an orbital shaker at 120 rpm (step 1). Following the logarithmic growth period, competitive co-culture was initiated by transferring 10% of AFP2 (48 h-old culture) and a 5% inoculum of C. neoformans (24 h-old culture) into 500 mL starch broth in a 1000 mL conical flask and growing them together at 37 °C over a 10 day period (step 2). The culture was serially passed every 10 days for 30 days. Whichever isolate displayed increased anticryptococcal activity in comparison with the parental strain was selected for each serial passage. AFP2 was then cultured alone using the quadrant streak technique on Actinomycetes isolation agar medium at 37 °C for 72 h. Different colonies with distinct morphologies were checked for anticryptococcal activity. The isolate that produced the largest ZOI (hereafter referred to as S1) was selected for a second round of adaptive competitive co-culture (step 3). The serial passages of AFP2 (S1) were continued until the third cycle of the adaptive co-culture experiment (steps 4–7). The best evolved strain from the third serial passage of the adaptive phase was selected that displayed higher antagonistic activity against C. neoformans than the parental and other evolved strains. However, we did not proceed to further serial passages as S3 exhibited suboptimal growth in comparison to the parental strain and it was also presumed that over time AFP2 would reach its maximum defense level against C. neoformans. During this study of selective pressure, we selected 3 potential evolved strains from each cycle of adaptive evolution, whichever displayed improved activity in comparison with the parental strain (Fig. 1). The evolved isolates S1, S2, and S3 were selected for further analysis of the production of secondary metabolites (step 8). The evolved strains were stored in 20% glycerol at −80 °C.26,28,43
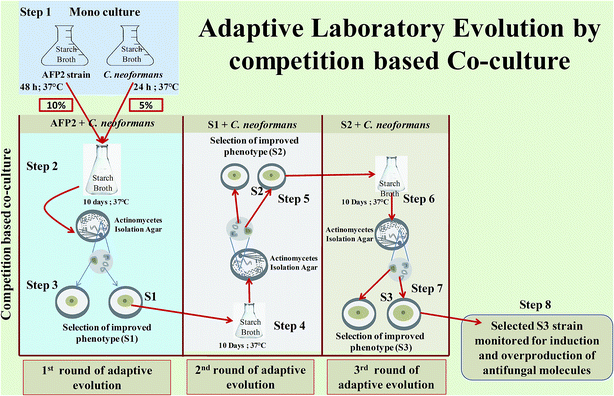 |
| | Fig. 1 Adaptive laboratory evolution by competition-based co-culture: step 1: monoculturing of AFP2 and C. neoformans for 48 h and 24 h, respectively; step 2: co-culturing of 10% AFP2 and 5% C. neoformans for 10 days; step 3: isolation of discrete colonies of AFP2 from co-culture broth and selection of phenotype with improved antifungal activity (evolved strain designated as S1); step 4: co-culturing of S1 and C. neoformans for a second round of ALE; step 5: isolation of discrete colonies of S1 from co-culture broth and selection of phenotype with improved antifungal activity (strain designated as S2); steps 6 and 7: the above procedure repeated for a third round of ALE; step 8: selection of potential evolved strain for monitoring the induction and overproduction of antifungal molecules in comparison with the parental strain. | |
Production and extraction of antifungal metabolites
A loopful of spores of parental-type S. variabilis AFP2, S1, S2, S3, and C. neoformans as a control were cultivated in 25 mL starch broth. The seed media were incubated at 37 °C for 48 h for the parental strain, S1 and S2, 72 h for S3 and 24 h for C. neoformans for the logarithmic growth period. Then, 10% (v/v) of each seed inoculum was used to seed 2.5 L production medium composed of yeast extract (0.1%), peptone (0.5%), glucose (1%), KH2PO4 (0.1%), and MgSO4 (0.05%) at 30 °C with a shaking speed of 180 rpm for an incubation period of 10 days. Each fermented broth was centrifuged at 8000g for 10 minutes. The culture filtrates were extracted twice with equal volumes of ethyl acetate using the cross-current method. Residual ethyl acetate was concentrated in vacuo to dryness. Finally, the extracts were dissolved in phosphate buffer (pH 7). The yield of each crude extract was observed and then the extracts were tested for antifungal activity by the well diffusion method, with a volume of 100 μL of the compound in each well.
Differential production of secondary metabolites by HPLC-UV-PDA
To monitor the differential production of secondary metabolites by the parental and evolved strains, ethyl acetate extracts of the parental strain, S1, S2 and S3 were analyzed using semi-preparatory HPLC (1260 Infinity, Agilent). A reverse-phase analytical C18 column (4.6 mm × 250 mm) was used with a linear gradient from 1% to 60% MeOH and 99% to 40% water over 30 minutes at a flow rate of 1 mL min−1 at 25 °C. For the first 4 minutes the MeOH concentration was increased linearly to 20%. At the twentieth minute, the MeOH concentration was increased linearly to 50%. Finally, the MeOH concentration was increased to 60% at 30 min. Chromatograms were obtained at different wavelengths (200 nm, 225 nm, 250 nm, and 300 nm) to study the possible metabolite profiles at various wavelengths.18
Antifungal activity of parental and S3 HPLC fractions
The HPLC fractions of the parental and S3 strains were collected using a peak-based method. The fractions were dried in an oven at 50 °C, dissolved in 50 μL PBS and tested for anticryptococcal activity (105 cells in each well) using a 96-well plate. Triplicates were measured for each fraction. The 96-well plate was incubated at 37 °C for 48 h in humid conditions to reduce evaporation of the media. The optical density of each well was recorded using an ELISA reader (Sunrise, Tecan) at 620 nm.15
Evaluation of evolution in Streptomyces variabilis AFP2
In general, morphological variants are evident in Streptomyces mutants.43 Hence, phenotypic changes in the parental and evolved strains were monitored by observing changes in colony and spore morphology. The spore morphology of 14 day-old culture samples of the parental strain and S3 strain was examined using light and scanning electron microscopy. The spore morphology was studied by examining gold-coated dehydrated specimens using a field emission scanning electron microscope (JSM-6701F, JEOL, Japan).
In addition, increases in the antagonistic activity of S3 were analyzed by culturing in the following conditions: (i) monoculture of the parental strain; (ii) monoculture of the S3 strain; (iii) monoculture of C. neoformans; (iv) co-culture of the parental strain and C. neoformans; and (v) co-culture of S3 and C. neoformans. The experiments were carried out for 14 days at 37 °C. Each day, 100 μL co-culture broth was withdrawn at time intervals of 24 h for 14 consecutive days and cell counts of C. neoformans were performed using a hemocytometer. In addition, changes in capsule size and the polysaccharide matrix that surrounded C. neoformans were assessed.
Measurement of capsule thickness by staining with India ink
A drop of India ink was mixed with an aliquot of C. neoformans on a glass slide. The samples were examined using a Nikon Eclipse Ci-L microscope and images were recorded with an SLR camera (Canon D5100). To calculate the relative size of the capsule, the diameter of the whole cell including the capsule (Dwc) and the diameter of the cell body limited by the cell wall (Dcb) were measured using ImageJ 1.48v software (National Institutes of Health, Washington, D.C.). The size of the capsule relative to that of the whole cell was defined as a percentage {[(Dwc − Dcb)/Dwc] × 100}. Eight cells were measured for each determination and the average was calculated.44–46
GC-MS analysis
A crude extract of the S3 strain was subjected to comparative analysis against an extract from the parental strain using gas chromatography and mass spectrometry (GC-MS) with the help of the National Institute of Standards and Technology spectral library.47 The analyses were performed using a PerkinElmer Clarus 500 GC-MS system. The program was set at a temperature of 50 °C for a duration of 1 min and the temperature was then raised at 10 °C min−1 to 150 °C (1 min hold), at 8 °C min−1 to 250 °C (1 min hold), and at 15 °C min−1 to 300 °C (3 min hold). Helium (1 mL min−1) was used as the carrier gas. The injector temperature was maintained at 280 °C and the mass range was 40–450 amu. A sample of 1 μL dissolved in ethanol was injected into the system. Compounds were identified by comparing their mass spectra with spectra from the database.
Results
Isolation of S. variabilis AFP2
During this study, 60 morphologically different Actinomycetes spp. from marine sediments were isolated using various agar media recommended for the isolation of Actinomycetes. However, only two isolates were shown to exhibit anticryptococcal activity, among which the AFP2 strain isolated from starch agar displayed a maximum ZOI of 12 mm. Thus, the AFP2 strain was selected for sequencing of 16S rRNA. A sequence similarity search using the BLAST tool revealed that AFP2 (KJ716228) belongs to a distinct phyletic line of S. variabilis. The isolate was closely related to the type strains of S. variabilis strain NRRL B-3984, S. labedae strain CSSP735, S. lateritius strain CSSP722 and S. griseoincarnatus strain CSSP407, sharing a homology of 99%.
Adaptive laboratory evolution by competition-based co-culture
ALE by competition-based co-culture was carried out to trigger the expression of anticryptococcal metabolites by AFP2. After serial passage for 30 days (∼180 generations) of AFP2 in competition-based co-culture with C. neoformans, three isolates, namely, S1, S2 and S3 were derived from a single evolved population in three adaptive cycles. During adaptive evolution, certain changes in the characteristics of S. variabilis are expected to occur. Hence, to confirm the putative effect, the evolved isolate from each adaptive phase was tested for anticryptococcal activity because the best phenotype is not necessarily the one with the highest ability to survive in competitive conditions, but the one that displays increased activity.28 The ZOIs of the evolved strains were comparatively larger than that of the parental strain, which suggests that antagonistic activity was higher in the evolved strains than in the parental strain. A similar kind of adaptive evolution was used by Charusanti et al. to increase the activity of S. clavuligerus against S. aureus; however, the experimental design was different from that in the present study.26,48,49
Increase in antifungal activity in evolved strains
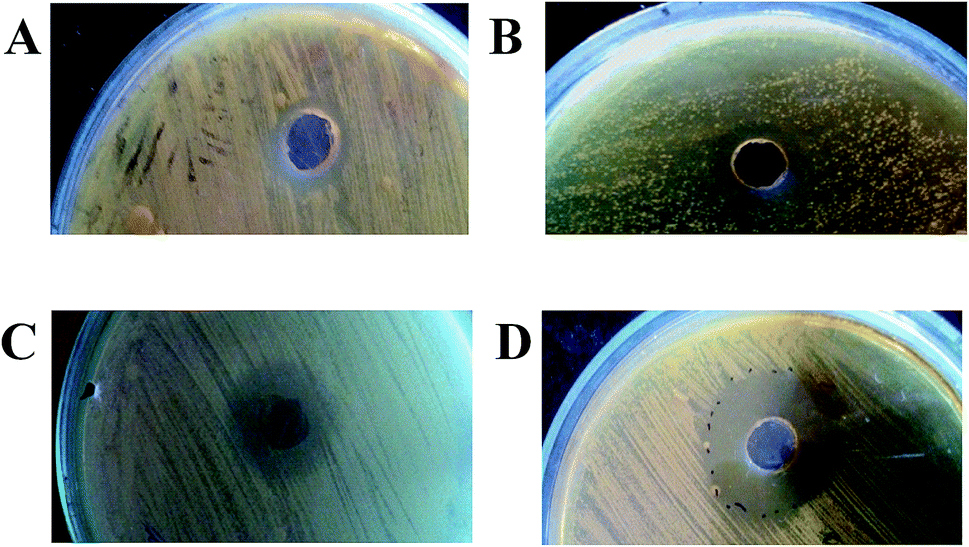
The parental-type, S1, S2, and S3 strains were cultivated in production medium for 10 days at 37 °C. The culture supernatants were then centrifuged and extracted with ethyl acetate. A variation in yield between the strains was observed (parental-type: 11.8 mg; S1 and S2: 12.6 mg; S3: 8.6 mg; and C. neoformans: 3 mg, for 100 mL production medium). A significant increase in antifungal activity for the crude extracts of the adapted strains is shown in Fig. 2: ZOI of parental strain: 12 mm; S1 and S2: 15 mm; and S3: 26 mm. Interestingly, the yield of compounds and the antagonistic activity were not directly proportional, which probably suggests that adaptation could most probably promote the synthesis of more specific inhibitory molecules than in the wild-type strain.
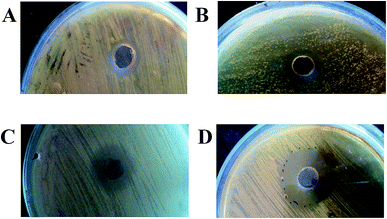 |
| | Fig. 2 Antagonism assay of EtOAc extracts of parental and evolved strains of AFP2 (A–D): ZOIs of parental, S1, S2, and S3 strains of AFP2. | |
Profiles of secondary metabolites by HPLC-UV-PDA
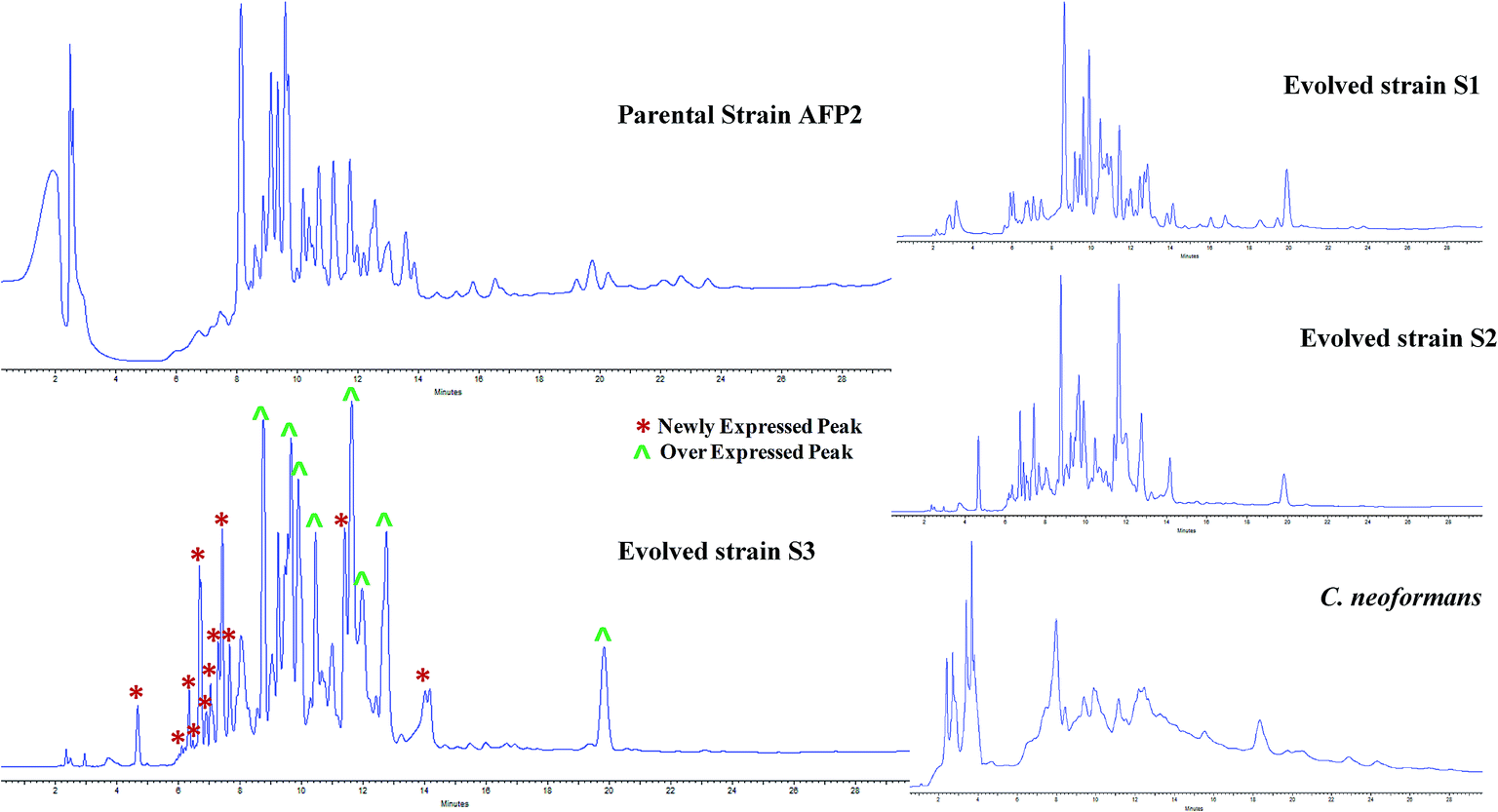
The profiles of secondary metabolites of the parental-type, S1, S2, and S3 strains were monitored by HPLC-UV-PDA. The maximum absorbance was observed at 225 nm for all the strains. The metabolomes of the evolved strains displayed a different chemotype than that of the parental-type strain (Fig. 3). A comparison of all the chromatograms implies that some new peaks were observed for S1, S2 and S3 between retention times of 4 and 8 minutes. In addition, the increase in peak area in the S3 chromatogram confirms that the triggered molecules and existing molecules were overexpressed. A comparison of the HPLC chromatograms of the parental and evolved strains suggests that progressive adaptation of strains over several passages is useful to trigger and promote the expression of metabolites. Many reports have demonstrated chemotypic differences in axenic and mixed cultures using HPLC-UV analysis;15,18,50 however, this is a presumptive test to confirm variations in the metabolome. Although the samples were analyzed at different wavelengths to screen the maximum number of peaks, it is mandatory to identify the chemotype of the parental and evolved strains.
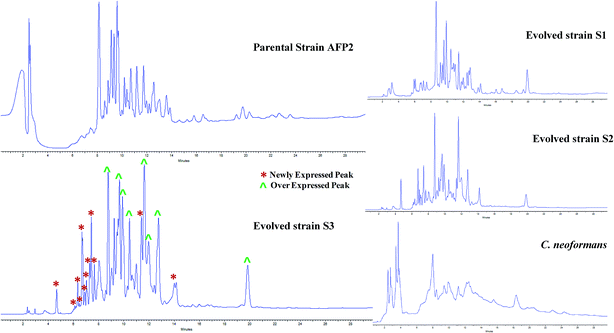 |
| | Fig. 3 Comparative study of HPLC profiles of EtOAc extracts of parental and evolved strains of AFP2. HPLC chromatograms of parental, S1, S2, and S3 strains of AFP2 and C. neoformans at 225 nm. In the S3 chromatogram (*) represents newly expressed peaks and (^) overexpressed peaks. | |
Antifungal activity of parental and S3 HPLC fractions
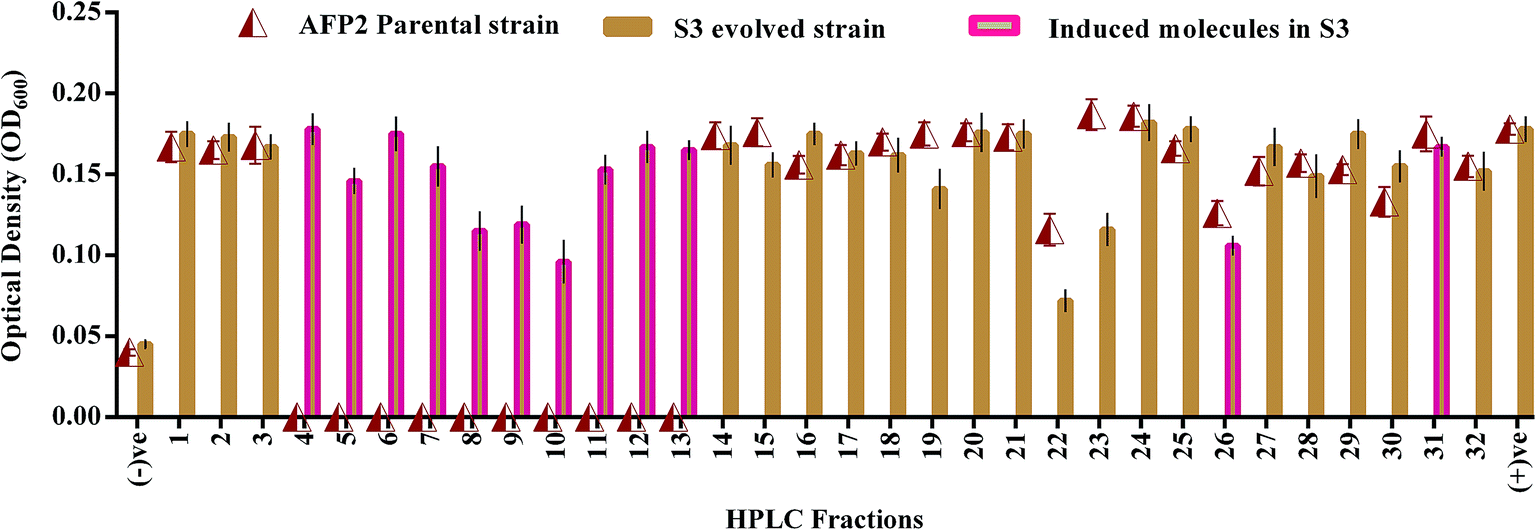
HPLC fractionation of the crude extracts of the wild-type and S3 strains yielded 22 and 32 fractions, respectively. Fig. 4 shows that nearly 12 molecules were newly expressed by the evolved strain. Among these, 3 fractions were shown to display antagonistic activities of 35–45% in terms of reduction in the growth of C. neoformans in comparison with the positive control. However, the 22nd fraction, which was found to be expressed by both parental-type and S3 strains, exhibited a 65% reduction in the growth of C. neoformans. Hence, the induction and overexpression of antifungal molecules were evident in the evolved S3 strain.
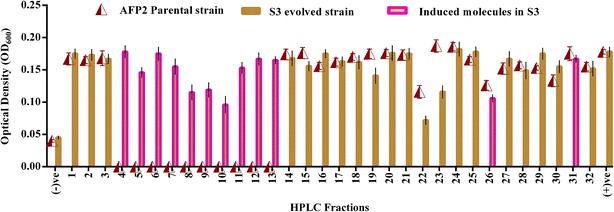 |
| | Fig. 4 Antifungal activity of HPLC fractions of parental and evolved S3 strains of AFP2. (−)ve represents negative control (media without culture); (+)ve represents positive control (media with culture). | |
Evaluation of evolution in AFP2
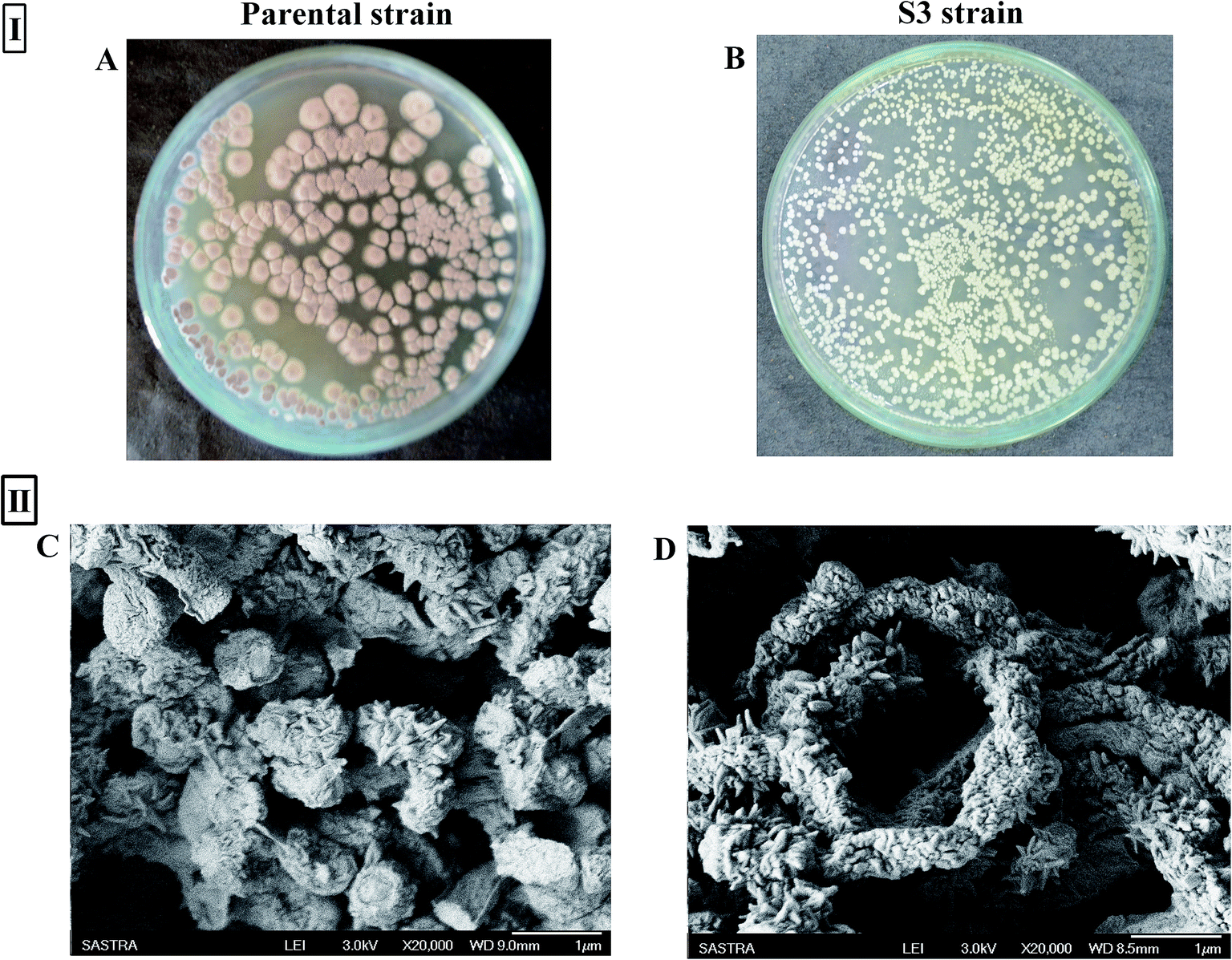
A notable phenotypic change in terms of colony morphology was observed in the S3 strain (Fig. 5I). Scanning electron microscopy analysis of 14 day-old spores of the parental strain showed the presence of wart-like projections and spines on spore surfaces. However, the S3 strain was characterized by tightly coiled spiral chains of spores, with spores having large numbers of long projections (Fig. 5II), unlike the somewhat straighter and loosely coiled chains of spores with fewer projections over the spore surface of the parent type.
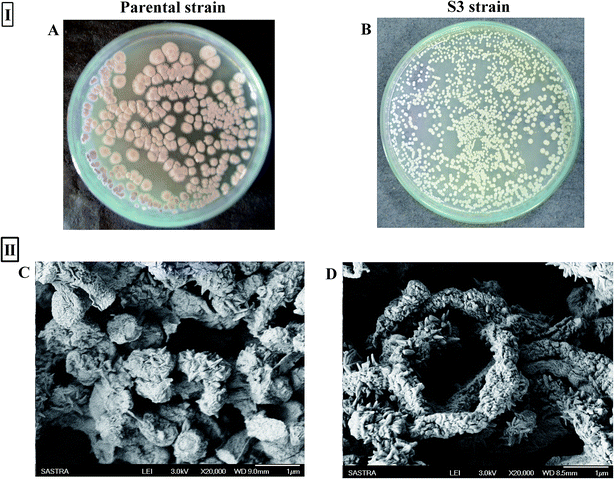 |
| | Fig. 5 (I) Variation between parental strain (A) and evolved strain (B) in colony morphology on AIA agar; (II) SEM micrographs of parental (C) and S3 (D) strains of AFP2. | |
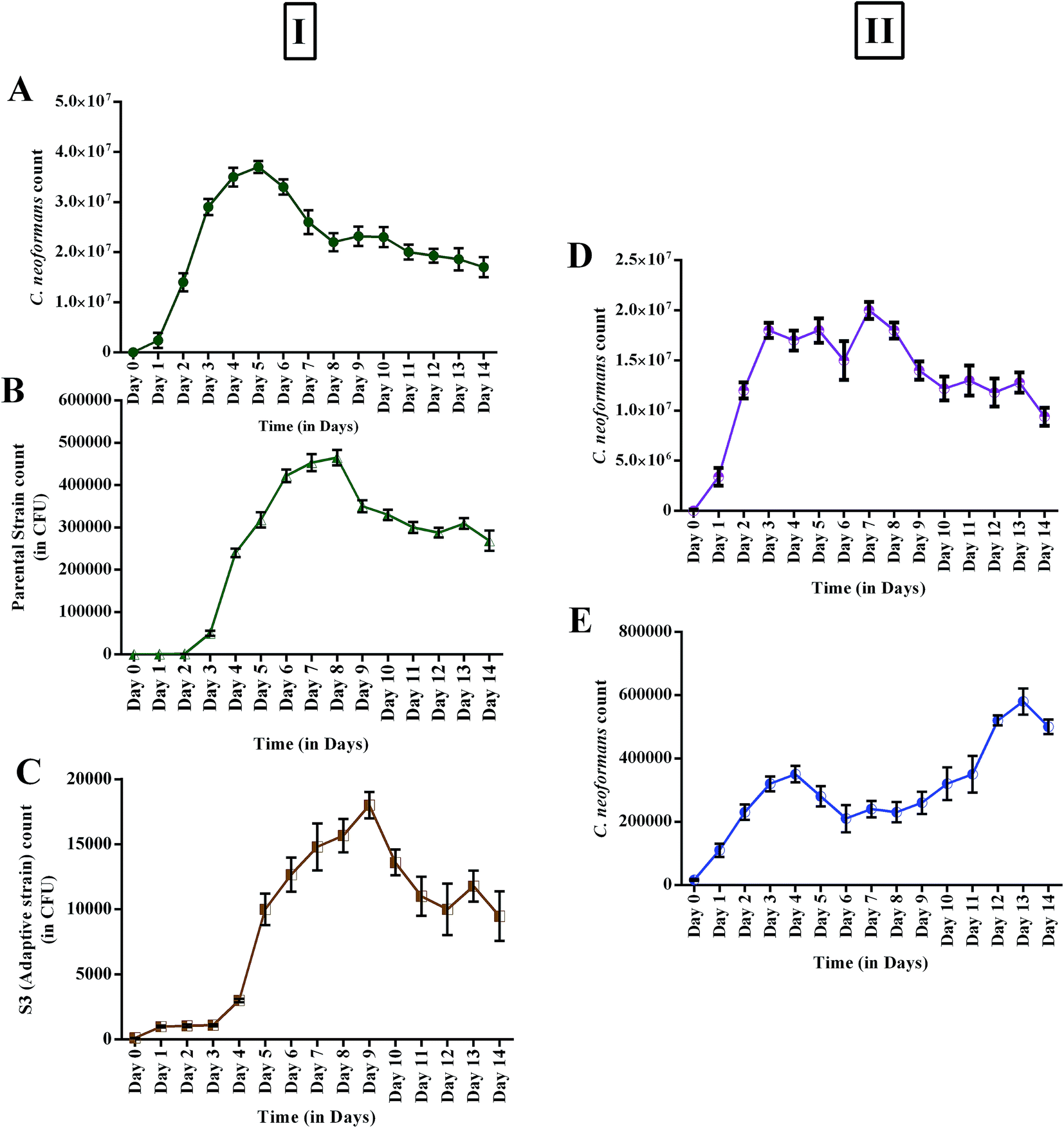
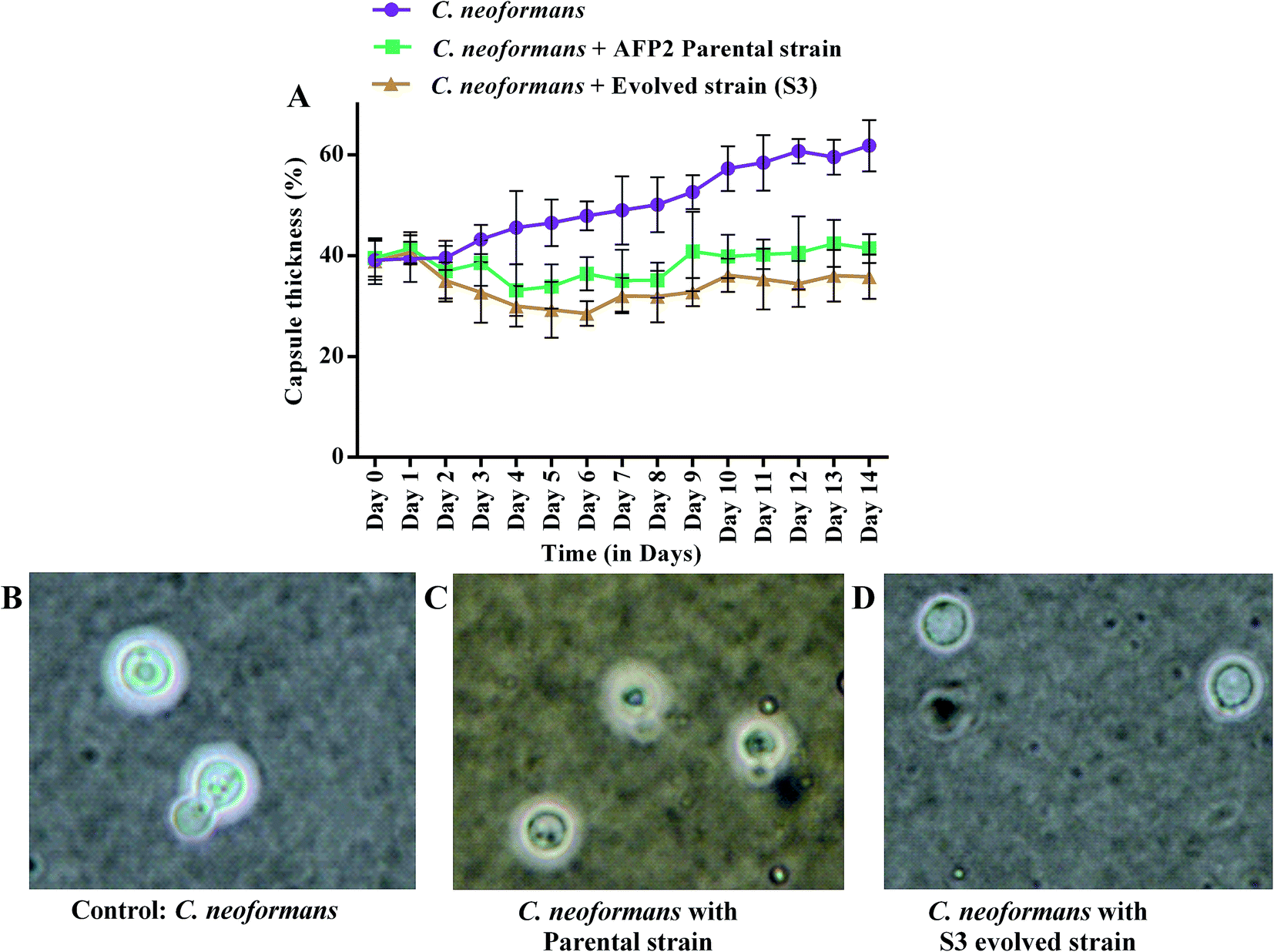
Changes in growth profile were also observed between the evolved and parental strains. The evolved strain displayed suboptimal growth when compared with the parental strain (Fig. 6I). The suboptimal growth of the S3 strain revealed that it underwent a competition for nutrients during co-cultivation. However, a high growth rate of a microbe is not a common criterion for determining evolutionary fitness.28,51 Nevertheless, biomass yield and an ability to survive in a competitive environment are always correlated with the production of secondary metabolites.52 Interestingly, increased activity was observed even for suboptimal growth of S3. Fig. 6II shows the reduction in the growth of C. neoformans during mixed culture with the parental strain. However, a remarkable reduction in fungal growth was observed for co-cultivation of S3 with C. neoformans. Moreover, a notable change in capsule thickness in the evolved strain is clearly evident in Fig. 7, with a reduction of 25% in capsule thickness.
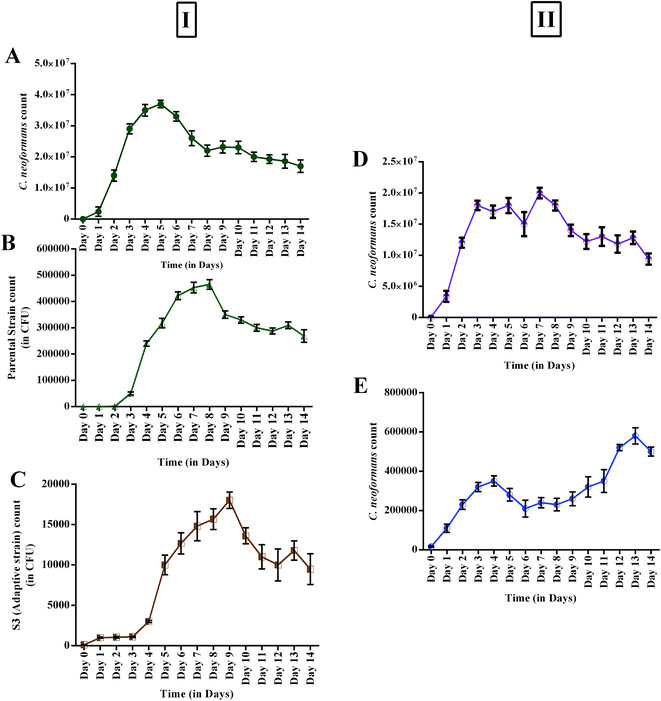 |
| | Fig. 6 (I) Growth profiles of monocultures (control): (A) C. neoformans; (B) AFP2 parental strain; (C) S3 evolved strain; (II) antagonistic effects of parental and evolved strains during co-culturing: (D) parental strain vs. C. neoformans; (E) S3 evolved strain vs. C. neoformans. | |
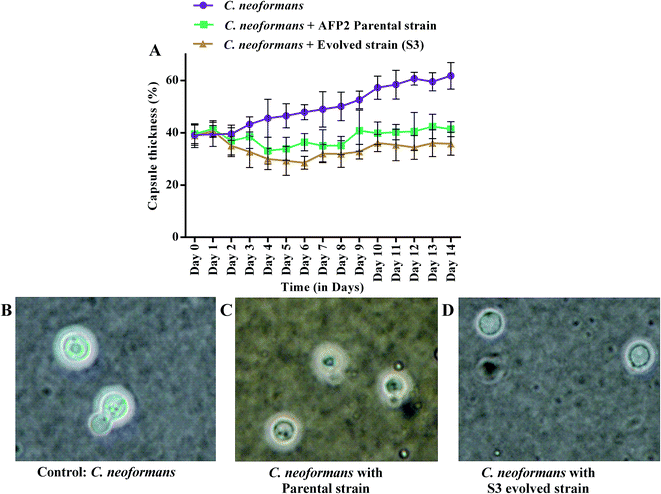 |
| | Fig. 7 Comparison of capsule growth of C. neoformans during co-cultivation with parental and S3 evolved strains: (A) the percentage capsule thickness for ≥8 cells per group is shown. The bars represent standard errors; (B–D) light microscopy views (100×) of C. neoformans capsules. | |
GC-MS analysis
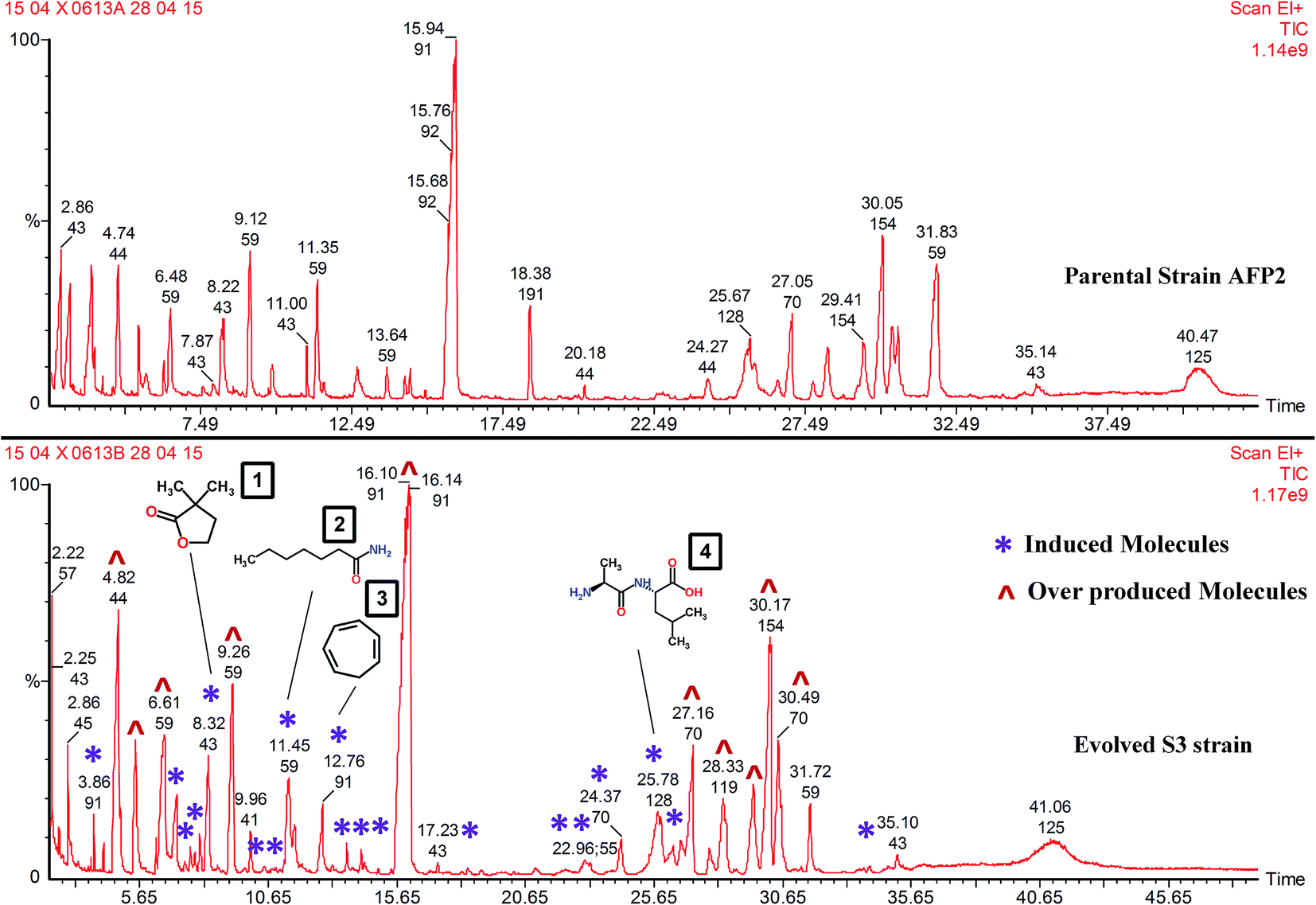
About 21 new metabolites alien to the parent strain were seen to be produced by S3 (Fig. 8). The list of compounds that are commonly expressed by the parental and S3 evolved strains is given in Table 1. The molecules triggered in S3 and those that are absent in the wild-type strain are listed in Table 2. In addition to interpreting the chemical diversity, the data for percentage peak area represents the yield of compounds. The increase in the yield of compounds is a notable result that proves that the integrated approach using ALE and co-cultivation is a suitable method for increasing the production of metabolite. For instance, increases in the yield of 2-methylpropanamide (56%), butanamide (54%), 3-methylbutanamide (53%) and hexahydro-3-(2-methylpropyl)-pyrrolo[1,2-a]pyrazine-1,4-dione (41%) were recorded in comparison with parental-type S. variabilis. Overproduction was clearly evident, as shown in Fig. 8 and Table 1. Few of the induced compounds and their derivatives were previously reported to have antifungal activity (Table 2). Nevertheless, molecules such as DL-alanyl-L-leucine (yield, % peak area: 3.1%), dihydro-3,3-dimethyl-2(3H)-furanone (2.7%), enanthamide (2.7%), 1,3,5-cycloheptatriene (1.6%) and 1-aziridineethanol (1.6%) were not reported to have antifungal activity. In addition, these molecules were not reported in S. variabilis. Hence, this confirms that our approach enables the triggering of silent gene cluster(s), resulting in the synthesis of novel molecules.
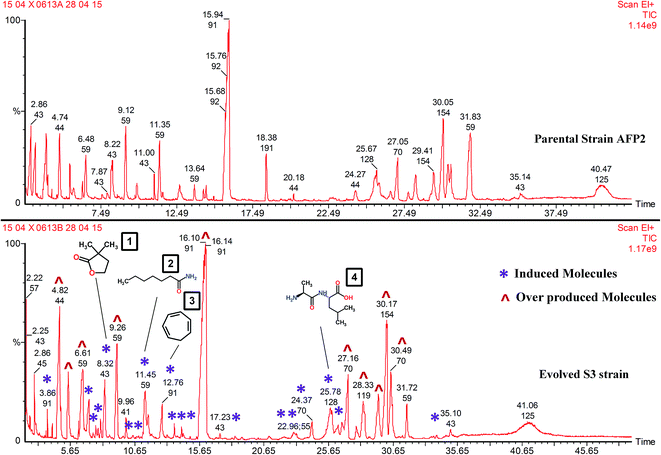 |
| | Fig. 8 GC-MS profiles of AFP2 parental and S3 evolved strains: in the S3 chromatogram (*) represents induced molecules and (^) represents overproduced molecules. (1) Dihydro-3,3-dimethyl-2(3H)-furanone; (2) enanthamide; (3) 1,3,5-cycloheptatriene; (4) DL-alanyl-L-leucine. | |
Table 1 Common compounds present in both wild-type strain and S3 strain determined by GC-MS
| S. no. |
Name |
Formula |
MW |
Wild-type |
S3 |
Overproduction of compound in S3 (% increase) |
| Retention time |
Peak area |
% peak area |
Retention time |
Peak area |
% peak area |
| 1 |
2-Methylpropanamide |
C4H9NO |
87 |
4.74 |
39873944 |
3.7718 |
4.82 |
1.39 × 108 |
8.6307 |
56.29787 |
| 2 |
Butanamide |
C4H9NO |
87 |
5.42 |
13945809 |
1.3192 |
5.48 |
46590024 |
2.8877 |
54.31658 |
| 3 |
N-Hydroxymethylacetamide |
C3H7NO2 |
89 |
7.87 |
4336799 |
0.4102 |
6.29 |
4697297 |
0.2911 |
— |
| 4 |
3-Methylbutanamide |
C5H11NO |
101 |
6.48 |
25926508 |
2.4525 |
6.61 |
85550336 |
5.3025 |
53.74823 |
| 5 |
Pentanamide |
C5H11NO |
101 |
9.12 |
43524428 |
4.1171 |
9.26 |
91229608 |
5.6545 |
27.18896 |
| 6 |
Octanamide |
C8H17NO |
143 |
13.64 |
8076171 |
0.7639 |
11.66 |
17206876 |
1.0665 |
28.37318 |
| 7 |
Octanamide |
C8H17NO |
143 |
— |
— |
— |
13.70 |
6632707 |
0.4111 |
— |
| 8 |
[(Methoxymethoxy)methyl]benzene |
C9H12O2 |
152 |
14.24 |
3329908 |
0.315 |
14.25 |
4154936 |
0.2575 |
— |
| 9 |
Tridecane |
C13H28 |
184 |
14.92 |
1007684 |
0.0953 |
14.92 |
696186 |
0.0432 |
— |
| 10 |
Benzeneacetamide |
C8H9NO |
135 |
15.76 |
2.85 × 108 |
26.9762 |
16.10 |
4.72 × 108 |
29.244 |
7.754753 |
| 11 |
2,4-Bis(1,1-dimethylethyl)phenol |
C14H22O |
206 |
18.38 |
24139044 |
2.2834 |
18.40 |
2209378 |
0.1369 |
— |
| 12 |
Tridecane |
C13H28 |
184 |
14.92 |
1007684 |
0.0953 |
14.92 |
696186 |
0.0432 |
— |
| 13 |
Tridecane |
C13H28 |
184 |
— |
— |
— |
20.19 |
608988 |
0.0377 |
— |
| 14 |
Hexahydro-3-(2-methylpropyl)-pyrrolo[1,2-a]pyrazine-1,4-dione |
C11H18N2O2 |
210 |
27.05 |
35255676 |
3.3349 |
27.16 |
67217472 |
4.1662 |
19.95343 |
| 15 |
4-Methylbenzoic acid 1-methylethyl ester |
C11H14O2 |
178 |
28.23 |
24750376 |
2.3412 |
28.33 |
51245500 |
3.1762 |
26.28928 |
| 16 |
Hexahydro-3-(2-methylpropyl)-pyrrolo[1,2-a]pyrazine-1,4-dione |
C11H18N2O2 |
210 |
29.41 |
21280488 |
2.013 |
29.52 |
55535760 |
3.4422 |
41.51996 |
| 17 |
Hexahydro-3-(2-methylpropyl)-pyrrolo[1,2-a]pyrazine-1,4-dione |
C11H18N2O2 |
210 |
30.05 |
79494712 |
7.5196 |
30.17 |
1.29 × 108 |
7.9727 |
5.683144 |
| 18 |
Hexahydro-3-(2-methylpropyl)-pyrrolo[1,2-a]pyrazine-1,4-dione |
C11H18N2O2 |
210 |
30.36 |
28916384 |
2.7353 |
30.48 |
67372280 |
4.1758 |
34.49638 |
| 19 |
Dodecanamide |
C12H25NO |
199 |
31.83 |
94955744 |
8.9821 |
34.00 |
2033383 |
0.126 |
— |
| 20 |
Hexahydro-3-(phenylmethyl)-pyrrolo[1,2-a]pyrazine-1,4-dione |
C14H16N2O2 |
244 |
40.47 |
72263544 |
6.8356 |
41.06 |
80100008 |
4.9647 |
— |
Table 2 Compounds present in S3 strain determined by GC-MS
| S. no. |
Name |
Formula |
MW |
Retention time |
Peak area |
% peak area |
| 1 |
1,3-Dimethylbenzene |
C8H10 |
106 |
3.86 |
4746478 |
0.2942 |
| 2 |
1-Aziridineethanol |
C4H9NO |
87 |
7.08 |
26188024 |
1.6232 |
| 3 |
1,1′-[Ethylidenebis(oxy)]bis[2-methylbutane] |
C12H26O2 |
202 |
7.39 |
1686723 |
0.1045 |
| 4 |
2-Methyl-3-butenoic acid methyl ester |
C6H10O2 |
114 |
7.6 |
3654044 |
0.2265 |
| 5 |
Butanedioic acid |
C4H6O4 |
118 |
13.12 |
1403604 |
0.087 |
| 6 |
4-Methylene-5-hexenal |
C7H10O |
110 |
7.79 |
2322845 |
0.144 |
| 7 |
Dihydro-3,3-dimethyl-2(3H)-furanone |
C6H10O2 |
114 |
8.32 |
44006676 |
2.7276 |
| 8 |
4-Acetylbutyric acid |
C6H10O3 |
130 |
10.49 |
2265020 |
0.1404 |
| 9 |
6-Methyl-6-azabicyclo[3.2.1]octane |
C8H15N |
125 |
10.69 |
196201 |
0.0122 |
| 10 |
Enanthamide |
C7H15NO |
129 |
11.45 |
44932220 |
2.7849 |
| 11 |
1,3,5-Cycloheptatriene |
C7H8 |
92 |
12.76 |
26539802 |
1.645 |
| 12 |
Dihydro-4,4-dimethyl-2(3H)-furanone |
C6H10O2 |
114 |
13.91 |
1851129 |
0.1147 |
| 13 |
Benzamide |
C7H7NO |
121 |
14.38 |
2886997 |
0.1789 |
| 14 |
2-Methyl-(4H)-3,1-benzoxazin-4-one |
C9H7NO2 |
161 |
15.43 |
698960 |
0.0433 |
| 15 |
Phenylpropanamide |
C9H11NO |
149 |
18.91 |
1882071 |
0.1167 |
| 16 |
4-Methylpentanamide |
C6H13NO |
115 |
21.04 |
3508549 |
0.2175 |
| 17 |
4-Hydroxybenzeneacetic acid |
C8H8O3 |
152 |
22.21 |
6275494 |
0.389 |
| 18 |
3-(1-Aziridinyl)-3-dimethylamino-2-propenal |
C7H12N2O |
140 |
24.37 |
14641184 |
0.9075 |
| 19 |
DL-Alanyl-L-leucine |
C9H18N2O3 |
202 |
25.78 |
51295148 |
3.1793 |
| 20 |
4-Hydroxyphenylacetamide |
C8H9NO2 |
151 |
26.4 |
8670816 |
0.5374 |
| 21 |
4-Ethyl-5-methylheptanamide |
C10H21NO |
171 |
26.68 |
14915393 |
0.9245 |
| 22 |
2,6,10,15-Tetramethylheptadecane |
C21H44 |
296 |
33.79 |
582825 |
0.0361 |
Evolutionary fitness analysis revealed that a 64-fold or 98% reduction in the growth of C. neoformans was observed for the evolved S3 strain. In addition, remarkable differences in the induction and overproduction of antifungal molecules were also evident for the S3 strain, with an increase of ∼52% in compounds and 21 new triggered compounds. Hence, the present study confirms that an integrated approach using adaptive laboratory techniques and co-culturing techniques is useful to discover potential molecules against targeted pathogens.
Discussion
Natural products from Actinomycetes spp. are considered to be an important source of novel antimicrobial compounds owing to their diversified secondary metabolites.53,54 Recently, the rate of discovery of new compounds from terrestrial Actinomycetes spp. has been decreasing, whereas the rate of rediscovery of known compounds has increased.55,56 As marine environmental conditions are extremely different from terrestrial conditions, it is surmised that marine Actinomycetes spp. have different characteristics from those of their terrestrial counterparts and therefore might produce different types of bioactive compounds. They form a stable, persistent population in various marine ecosystems.57 There have been some recent discoveries of new antifungal molecules, namely, the polyene-polyol-macrolides bahamaolide A and B,58 ikarugamycin derivatives from marine S. variabilis59–61 and a new tetramic acid glycoside, aurantoside K, obtained from a marine sponge.62,63 In addition, more information was provided by Xu et al. in a recent review, which reports 116 new antifungal and antibacterial compounds from marine fungi during 2010-15.64 Many studies propose the existence of undiscovered antifungal compounds from marine Actinomycetes spp. The present study exploited marine sediments for Actinomycetes spp. exhibiting anticryptococcal activity. During the course of this study, we found a new strain of S. variabilis AFP2 that has an inhibitory effect against C. neoformans, which suggests the production of possible new antifungal metabolites. However, the chances of rediscovering the tens of thousands of known molecules are high. The main challenges behind this are the inability to cultivate potential microbes and finding a suitable trigger that enables the expression of silent biosynthetic gene clusters under normal laboratory conditions. Novel approaches are being implemented by various researchers to search for antimicrobial agents such as genome mining and soil metagenomics.43,48,65 Until now various co-culture strategies have been employed to mimic natural conditions to facilitate the discovery of novel secondary metabolites.66,67 The mixed cultivation of microbes has drawn the attention of most investigators, as the interspecies communication and defense mechanisms are found to trigger the expression of silent biosynthetic gene clusters.68,69 However, mixed fermentation is still in its infancy, which is probably due to lack of reproducibility.70 More recently, two new butyrolactone derivatives were isolated during the co-cultivation of A. terreus with the bacteria Bacillus subtilis and Bacillus cereus.71 Similarly, a new molecule, rhodostreptomycin, was expressed during the co-cultivation of Rhodococcus fascians and S. padanus,72 which was not detected in axenic cultures. However, co-culture may not always be suitable for discovering novel molecules. For example, levorin (an existing molecule) was isolated during co-cultivation of Candida tropicalis and Actinomyces levoris.73,74 Similarly, 6-methylsalicylic acid and cyclo-(phe–phe) dipeptides were isolated during the co-cultivation of two unknown marine endophytic fungi.75 Also, in a recent study, co-cultivation of Aspergillus fumigatus and S. bullii was reported to produce seven metabolites belonging to the diketopiperazine alkaloid family with antimicrobial and antiprotozoan activity.21 However, significant numbers of known compounds were reported and thus mixed fermentation is not always likely to yield novel molecules.76 Hence, until now various co-culture strategies have been employed to mimic natural conditions to facilitate the discovery of novel secondary metabolites.67 In the present study, we followed a different approach by combining adaptive laboratory evolution and mixed fermentation. Our idea was to evolve improved phenotypes that provide an efficient defense mechanism against the targeted pathogen by the overexpression and induction of novel molecules. In general, ALE involves the cultivation of microbes in controlled conditions for prolonged periods in the range of weeks to years, which enables the selection of improved phenotypes. Evolutionary engineering has been proven to be an efficient method for the improvement of industrial strains. This technique has been successfully established to generate strains with improved activity such as tolerance to thermal stress, nutrient stress, osmotic stress, ethanol stress, and acid stress and also for enhanced substrate utilization.77–79 The primary motive of the present study was to apply the evolutionary principle to generate improved antagonistic properties via possible methods for activating silent gene clusters by co-cultivation. During our study, by adaptive evolution and the competitive co-culture of S. variabilis AFP2 and C. neoformans for periods of 30 days, we have selected 3 improved phenotypes with higher anticryptococcal activity and their metabolomes were analysed.
In general, the common analytical techniques for the analysis of metabolomes include LC-MS, GC-MS and NMR. GC-MS is recognized to be one of the most versatile analytical methods in metabolomics studies of biological samples, because of its reliable and reproducible results.80,81 However, reports on microbial metabolomics using GC-MS are limited.82 Cevallos-Cevallos et al. demonstrated the suitability of GC-MS for microbial metabolomics of E. coli, Bacillus sp. and Pseudomonas sp. More frequently, researchers have used HPLC/LC-MS followed by NMR to determine the structural details of triggered molecules.21,71,83 However, as our intention was to compare the metabolic profiles of the parental and evolved strains, a GC-MS platform was used. GC-MS analysis showed clear comparisons between the metabolomes of the parental and S3 strains with respect to peak identification, peak relative area, retention time and mass spectroscopic data (Fig. 8). Structural determination using NMR is a future direction of our lab. Although GC-MS analysis determines the overexpression and induction of compounds, we believe that biological assays are also mandatory. Therefore, we tested the extracts of the parental, S1, S2 and S3 strains against C. neoformans and its capsule growth. The capsule is the most prominent and well-studied antiphagocytic factor in C. neoformans.84,85 The efficiency of phagocytosis is regulated by the capsule size. An increase in capsule size is found to decrease complement-mediated phagocytosis.86,87 Hence, the determination of the in vitro activity of the extracts of the parental and evolved strains and its influence on the capsule size of C. neoformans was considered to be another significant result of the present study, which is clearly depicted in Fig. 7. The evolved strain displayed a 64-fold or 98% reduction in the growth of C. neoformans in comparison with the parental strain. The increased antifungal activity was possibly due to the induction of new molecules and overexpression of compounds in the S3 strain. GC-MS analysis revealed an increase of ∼52% in the production of metabolites. Hence, the present study concludes with evidence that adaptive laboratory evolution by competition-based co-culture is a novel and suitable method of strain engineering to induce and overproduce specific molecules against a targeted pathogen.
Conclusion
The main challenge behind the discovery of novel anti-infective agents from microbial sources is to find a suitable trigger that enables the expression of silent biosynthetic gene clusters under normal laboratory conditions. Novel approaches are being implemented by various researchers to discover novel antimicrobial agents such as genome mining, soil metagenomics and co-culture techniques employed to mimic natural conditions to facilitate the discovery of novel secondary metabolites. However, co-culture may not always be suitable for discovering novel molecules. Our results suggests that the proposed integrated approach using adaptive laboratory evolution and competition-based co-culture is a novel and suitable method of strain engineering to induce and overproduce specific molecules against a targeted pathogen.
Conflict of interest
The authors declare no conflict of interests.
Funding
Department of Science & Technology (DST), New Delhi, under the Fast Track Young Scientist Scheme (No. SB/FT/LS-249/2012) and EMR scheme SR/S0/HS-0073/2012.
Acknowledgements
This study was financially supported by the Science and Engineering Research Board (SERB), Department of Science & Technology, New Delhi, under the Fast Track Young Scientist Scheme (No. SB/FT/LS-249/2012) and HPLC facility EMR scheme SR/S0/HS-0073/2012 to JP. We thank SASTRA University for providing us with the infrastructure to carry out our research work.
References
- M. A. Pfaller, P. G. Pappas and J. R. Wingard, Clin. Infect. Dis., 2006, 43, S3–S14 CrossRef CAS.
- C.-Y. Low and C. Rotstein, F1000 Med. Rep., 2011, 3, 1–8 Search PubMed.
- L. Pagano, M. Akova, G. Dimopoulos, R. Herbrecht, L. Drgona and N. Blijlevens, J. Antimicrob. Chemother., 2011, 66, 5–14 CrossRef PubMed.
- J. A. Vazquez, M. H. Miceli and G. Alangaden, Ther. Adv. Infect. Dis., 2013, 1, 85–105 Search PubMed.
- S. Antinori, ISRN AIDS, 2013, 1–22 CrossRef PubMed.
- D. J. Sloan and V. Parris, J. Clin. Epidemiol., 2014, 169–182 CrossRef PubMed.
- S. Bertozzi, N. S. Padian, J. Wegbreit, L. M. DeMaria, B. Feldman, H. Gayle, J. Gold, R. Grant and M. T. Isbell, in Disease control priorities in developing countries, World Bank, 2006, pp. 331–370 Search PubMed.
- J. Ramakrishnan, S. S. Rathore and T. Raman, RSC Adv., 2016, 6(48), 42387–42401 RSC.
- B. J. Park, K. A. Wannemuehler, B. J. Marston, N. Govender, P. G. Pappas and T. M. Chiller, AIDS, 2009, 23, 525–530 CrossRef PubMed.
- A. Loyse, D. Wilson, G. Meintjes, J. N. Jarvis, T. Bicanic, L. Bishop, K. Rebe, A. Williams, S. Jaffar, L. G. Bekker, R. Wood and T. S. Harrison, Clin. Infect. Dis., 2012, 54, 121–128 CrossRef CAS PubMed.
- J. A. Vazquez, HIV AIDS Res. Palliat. Care, 2010, 89–101 CrossRef CAS.
- B. V. Gandhi, M. M. Bahadur, H. Dodeja, V. Aggrwal, A. Thamba and M. Mali, J. Postgrad. Med., 2005, 51, 30–36 Search PubMed.
- M. F. Vicente, A. Basilio, A. Cabello and F. Peláez, Clin. Microbiol. Infect., 2003, 9, 15–32 CrossRef CAS PubMed.
- A. L. Demain and S. Sanchez, J. Antibiot., 2009, 62, 5–16 CrossRef CAS PubMed.
- Y. Dashti, T. Grkovic, U. R. Abdelmohsen, U. Hentschel and R. J. Quinn, Mar. Drugs, 2014, 12, 3046–3059 CrossRef PubMed.
- Y. M. Chiang, S. L. Chang, B. R. Oakley and C. C. C. Wang, Curr. Opin. Chem. Biol., 2011, 137–143 CrossRef CAS PubMed.
- Y.-M. Chiang, S.-L. Chang, B. R. Oakley and C. C. C. Wang, Curr. Opin. Chem. Biol., 2011, 15, 137–143 CrossRef CAS PubMed.
- V. Schroeckh, K. Scherlach, H.-W. Nützmann, E. Shelest, W. Schmidt-Heck, J. Schuemann, K. Martin, C. Hertweck and A. A. Brakhage, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 14558–14563 CrossRef CAS PubMed.
- U. R. Abdelmohsen, T. Grkovic, S. Balasubramanian, M. S. Kamel, R. J. Quinn and U. Hentschel, Biotechnol. Adv., 2015, 33, 798–811 CrossRef CAS PubMed.
- K. J. K. Luti and F. Mavituna, Appl. Microbiol. Biotechnol., 2011, 90, 461–466 CrossRef CAS PubMed.
- M. E. Rateb, I. Hallyburton, W. E. Houssen, A. T. Bull, M. Goodfellow, R. Santhanam, M. Jaspars and R. Ebel, RSC Adv., 2013, 3, 14444 RSC.
- A. Marmann, A. H. Aly, W. Lin, B. Wang and P. Proksch, Mar. Drugs, 2014, 12, 1043–1065 CrossRef PubMed.
- R. K. Pettit, Microb. Biotechnol., 2011, 471–478 CrossRef CAS PubMed.
- A. Vasconsuelo and R. Boland, Plant Sci., 2007, 172, 861–875 CrossRef CAS.
- L. Goers, P. Freemont and K. M. Polizzi, J. R. Soc., Interface, 2014, 11(96), 20140065 CrossRef PubMed.
- P. Charusanti, N. L. Fong, H. Nagarajan, A. R. Pereira, H. J. Li, E. A. Abate, Y. Su, W. H. Gerwick and B. O. Palsson, PLoS One, 2012, 7, 1–12 Search PubMed.
- I. Stergiopoulos, J. Collemare, R. Mehrabi and P. J. G. M. De Wit, FEMS Microbiol. Rev., 2013, 67–93 CrossRef CAS PubMed.
- M. Dragosits and D. Mattanovich, Microb. Cell Fact., 2013, 12, 1–17 CrossRef PubMed.
- Y. Wang, R. Manow, C. Finan, J. Wang, E. Garza and S. Zhou, J. Ind. Microbiol. Biotechnol., 2011, 38, 1371–1377 CrossRef CAS PubMed.
- D. Stanley, S. Fraser, P. J. Chambers, P. Rogers and G. A. Stanley, J. Ind. Microbiol. Biotechnol., 2010, 37, 139–149 CrossRef CAS PubMed.
- T. Horinouchi, K. Tamaoka, C. Furusawa, N. Ono, S. Suzuki, T. Hirasawa, T. Yomo and H. Shimizu, BMC Genomics, 2010, 11, 579 CrossRef PubMed.
- E. Kuster and S. T. Williams, Nature, 1964, 202, 928–929 CrossRef.
- M. A. Pfaller and D. J. Diekema, J. Clin. Microbiol., 2004, 42, 4419–4431 CrossRef CAS PubMed.
- H. S. Chaudhary, J. Yadav, A. R. Shrivastava, S. Singh, A. K. Singh and N. Gopalan, J. Adv. Pharm. Technol. Res., 2013, 4, 118–123 CrossRef PubMed.
- B. M. Rajan and K. Kannabiran, Int. J. Mol. Cell. Med., 2014, 3, 130–137 CAS.
- G. P. Eccleston, P. R. Brooks and D. I. Kurtboke, Mar. Drugs, 2008, 6, 243–261 CrossRef CAS PubMed.
- K. Selvam, B. Vishnupriya and V. S. C. Bose, Int. J. Pharm. Biol. Arch., 2011, 2, 1481–1487 Search PubMed.
- N. Shojaee, B. G. H. Shahidi, S. Shahdaei and S. B. Leyla, Afr. J. Microbiol. Res., 2014, 8, 1501–1509 CrossRef.
- J. Ramakrishnan, M. Shunmugasundaram and M. Narayanan, Iran. J. Biotechnol., 2009, 7, 75–81 CAS.
- M. Kimura, J. Mol. Evol., 1980, 16, 111–120 CrossRef CAS PubMed.
- A. J. Abid and S. A. Hamza, Sky J. Microbiol. Res., 2014, 2, 37–40 Search PubMed.
- K. Meesap, N. Boonapatcharoen, S. Techkarnjanaruk and P. Chaiprasert, J. Biomed. Biotechnol., 2012, 1–11 CrossRef PubMed.
- D. J. N. Gordon and M. Cragg, Appl. Environ. Microbiol., 2015, 81, 2284–2298 CrossRef PubMed.
- S. Frases, B. Pontes, L. Nimrichter, M. L. Rodrigues, N. B. Viana and A. Casadevall, Biophys. J., 2009, 97, 937–945 CrossRef CAS PubMed.
- R. García-Rodas, R. J. B. Cordero, N. Trevijano-Contador, G. Janbon, F. Moyrand, A. Casadevall and O. Zaragoza, mBio, 2014, 5, 1–13 CrossRef PubMed.
- S. S. Rathore, T. Raman and J. Ramakrishnan, Front. Microbiol., 2016, 7, 1–16 Search PubMed.
- S. E. Stein, NIST Standard Reference Database 1A v14, Natl. Inst. Stand. Technol. is an agency U.S. Dep. Commer., 2014 Search PubMed.
- J. Davies and D. Davies, Microbiol. Mol. Biol. Rev., 2010, 74, 417–433 CrossRef CAS PubMed.
- L. Xu, F. Wang, Y. Shen, H. Hou, W. Liu, C. Liu, C. Jian, Y. Wang, M. Sun and Z. Sun, Exp. Ther. Med., 2014, 7, 1516–1520 Search PubMed.
- M. E. Rateb, I. Hallyburton, W. E. Houssen, A. T. Bull, M. Goodfellow, R. Santhanam, M. Jaspars and R. Ebel, RSC Adv., 2013, 3, 14444 RSC.
- K. Vetsigian, R. Jajoo and R. Kishony, PLoS Biol., 2011, 9, e1001184 CAS.
- R. K. Bajpai and M. Reuss, Biotechnol. Bioeng., 1981, 23, 717–738 CrossRef CAS.
- J. F. Imhoff, A. Labes and J. Wiese, Biotechnol. Adv., 2011, 29, 468–482 CrossRef CAS PubMed.
- S. B. Zotchev, J. Biotechnol., 2012, 158, 168–175 CrossRef CAS PubMed.
- D. B. Xu, W. W. Ye, Y. Han, Z. X. Deng and K. Hong, Mar. Drugs, 2014, 12, 2590–2613 CrossRef CAS PubMed.
- Y. Song, G. Liu, J. Li, H. Huang, X. Zhang, H. Zhang and J. Ju, Mar. Drugs, 2015, 13, 1304–1316 CrossRef CAS PubMed.
- J. Prudhomme, E. McDaniel, N. Ponts, S. Bertani, W. Fenical, P. Jensen and K. Le Roch, PLoS One, 2008, 3, 1–8 Search PubMed.
- D. G. Kim, K. Moon, S. H. Kim, S. H. Park, S. Park, S. K. Lee, K. B. Oh, J. Shin and D. C. Oh, J. Nat. Prod., 2012, 75, 959–967 CrossRef CAS PubMed.
- R. Lacret, D. Oves-Costales, C. Gómez, C. Díaz, M. de la Cruz, I. Pérez-Victoria, F. Vicente, O. Genilloud and F. Reyes, Mar. Drugs, 2015, 13, 128–140 CrossRef CAS PubMed.
- C. E. Salomon, N. A. Magarvey and D. H. Sherman, Nat. Prod. Rep., 2004, 21, 105–121 RSC.
- K. Kyeremeh, K. S. Acquah, A. Sazak, W. Houssen, J. Tabudravu, H. Deng and M. Jaspars, Mar. Drugs, 2014, 12, 999–1012 CrossRef PubMed.
- C. Y. Wang, B. G. Wang, S. Wiryowidagdo, V. Wray, R. Van Soest, K. G. Steube, H. S. Guan, P. Proksch and R. Ebel, J. Nat. Prod., 2003, 66, 51–56 CrossRef CAS PubMed.
- R. Kumar, R. Subramani, K. D. Feussner and W. Aalbersberg, Mar. Drugs, 2012, 10, 200–208 CrossRef CAS PubMed.
- L. Xu, W. Meng, C. Cao, J. Wang, W. Shan and Q. Wang, Mar. Drugs, 2015, 13, 3479–3513 CrossRef CAS PubMed.
- L. Zhang, Integrated approaches for discovering novel drugs from microbial natural products, 2005 Search PubMed.
- H. Zhang, B. Pereira, Z. Li and G. Stephanopoulos, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 8266–8271 CrossRef CAS PubMed.
- J. D. van Elsas, A. V. Semenov, R. Costa and J. T. Trevors, ISME J., 2011, 5, 173–183 CrossRef PubMed.
- S. Roier, F. G. Zingl, F. Cakar, S. Durakovic, P. Kohl, T. O. Eichmann, L. Klug, B. Gadermaier, K. Weinzerl, R. Prassl, A. Lass, G. Daum, J. Reidl, M. F. Feldman and S. Schild, Nat. Commun., 2016, 7, 10515 CrossRef CAS PubMed.
- T. Netzker, J. Fischer, J. Weber, D. J. Mattern, C. C. Konig, V. Valiante, V. Schroeckh and A. A. Brakhage, Front. Microbiol., 2015, 6, 1–13 Search PubMed.
- J. Bader, E. Mast-Gerlach, M. K. Popović, R. Bajpai and U. Stahl, J. Appl. Microbiol., 2010, 109, 371–387 CrossRef CAS PubMed.
- H. Chen, G. Daletos, M. S. Abdel-Aziz, D. Thomy, H. Dai, H. Brotz-Oesterhelt, W. Lin and P. Proksch, Phytochem. Lett., 2015, 12, 35–41 CrossRef CAS.
- K. Kurosawa, I. Ghiviriga, T. G. Sambandan, P. A. Lessard, J. E. Barbara, C. Rha and A. J. Sinskey, J. Am. Chem. Soc., 2008, 130, 1126–1127 CrossRef CAS PubMed.
- E. P. Iakovleva and E. N. Sokolova, Antibiotiki, 1978, 23, 199–203 CAS.
- R. K. Pettit, Appl. Microbiol. Biotechnol., 2009, 83, 19–25 CrossRef CAS PubMed.
- F. Zhu, Y. Lin and J. Ding, Chem. Ind. For. Prod., 2007, 27, 8–10 CAS.
- A. Marmann, A. H. Aly, W. Lin, B. Wang and P. Proksch, Mar. Drugs, 2014, 12, 1043–1065 CrossRef PubMed.
- S. Oide, W. Gunji, Y. Moteki, S. Yamamoto, M. Suda, T. Jojima, H. Yukawa and M. Inui, Appl. Environ. Microbiol., 2015, 81, 2284–2298 CrossRef CAS PubMed.
- J. Winkler, L. H. Reyes and K. C. Kao, Adaptive Laboratory Evolution for Strain Engineering, 2013, vol. 985 Search PubMed.
- R. A. LaCroix, T. E. Sandberg, E. J. O'Brien, J. Utrilla, A. Ebrahim, G. I. Guzman, R. Szubin, B. O. Palsson and A. M. Feist, Appl. Environ. Microbiol., 2015, 81, 17–30 CrossRef PubMed.
- O. Fiehn, Comp. Funct. Genomics, 2001, 2, 155–168 CrossRef CAS PubMed.
- H. Tsugawa, Y. Tsujimoto, M. Arita, T. Bamba and E. Fukusaki, BMC Bioinf., 2011, 12, 131 CrossRef CAS PubMed.
- L. Miao, T. F. N. Kwong and P. Y. Qian, Appl. Microbiol. Biotechnol., 2006, 72, 1063–1073 CrossRef CAS PubMed.
- K. M. Zuck, S. Shipley and D. J. Newman, J. Nat. Prod., 2011, 74, 1653–1657 CrossRef CAS PubMed.
- O. W. Liu, C. D. Chun, E. D. Chow, C. Chen, H. D. Madhani and S. M. Noble, Cell, 2008, 135, 174–188 CrossRef CAS PubMed.
- C. D. Chun, J. C. S. Brown and H. D. Madhani, Cell Host Microbe, 2011, 9, 243–251 CAS.
- C. P. Taborda and A. Casadevall, Immunity, 2002, 16, 791–802 CrossRef CAS PubMed.
- O. Zaragoza, C. P. Taborda and A. Casadevall, Eur. J. Immunol., 2003, 33, 1957–1967 CrossRef CAS PubMed.
Footnote |
| † All authors have equal contribution. |
|
| This journal is © The Royal Society of Chemistry 2016 |
Click here to see how this site uses Cookies. View our privacy policy here.