
Association behaviors of carbazole-labeled polyacrylamide in water studied by fluorescence spectroscopy†
Yongjun Guo*ab,
Chao Zheng*c,
Hongmei Yangc and
Yan Liangc
aState Key Laboratory of Oil and Gas Reservoir Geology and Exploitation, Southwest Petroleum University, Chengdu 610500, People's Republic of China. E-mail: gyfzgyj@126.com
bSchool of Chemistry and Chemical Engineering, Southwest Petroleum University, Chengdu 610500, People's Republic of China
cDepartment of Research & Development, Sichuan Guangya Polymer Chemical Company, Limited, Chengdu 610500, People's Republic of China. E-mail: zcphenix@163.com
First published on 1st September 2016
Abstract
In this work association behaviors of a carbazole-labeled hydrophobically modified polyacrylamide were studied by fluorescence spectroscopy in order to reveal the aggregation-induced spectral features. Both emission and excitation spectra reveal three distinct regimes for the association process of carbazole-labeled polyacrylamide with increasing concentration. In regime I, there is only a linear relationship between fluorescence intensity and concentration which was ascribed to unimer regime without aggregation. While a marked change in spectral shape could be observed in regimes II and III beyond the critical aggregation concentration. In regime II there is an aggregation-induced red shift in the excitation spectra and a decrease in 0–0 transition in emission spectra. In regime III aggregation-induced quenching was observed. These spectroscopic features of the excitation and emission spectra imply the formation of disordered J-aggregates by carbazole units during the association process, which could be employed to analyse the structural transition of hydrophobically modified polyacrylamide solutions.
Introduction
Hydrophobically modified water-soluble polymer (HMWSP) is a kind of polymer with a small amount (<2%) of hydrophobic units introduced to the hydrophilic backbone, which is of great importance in industries such as food1 and coatings2 as well as enhanced oil recovery.3,4 Its most important solution structure is associative networks resulting from the aggregation of hydrophobic units, when the concentration is above a critical value.5,6 Thus the association of HMWSP in water determines its performance in various applications. Due to the high viscosity of HMWSP solutions, characterization of the association behaviours of HMWSPs in water is not as easy as that for common block copolymer systems because electron microscopy is limited by the difficulty in sample preparation. For this reason fluorescence spectroscopy becomes one of the most powerful tools to study the hydrophobic micro-domains according to either the sensitivity of chromophores to polarity7–9 or a diffusion controlled process: excimer formation.7,10–13 In most relevant research, pyrene and its analogues were adopted as labeled molecules because of their well-known fluorescence characteristics.14 As to the emission spectrum of pyrene, the ratio I1/I3 (the intensities of the first and third vibronic peaks) is a sensitive indicator of the polarity of the pyrene microenvironment, which was widely used to detect micelle formation resulting from change in pH,7 concentration9 and so forth. In addition, relative excimer emission IE/IM has also been introduced to study the influence of polymer concentration12,13 or surfactant15 on hydrophobic aggregates.However, multistep synthesis was required of pyrene-labeled monomer design for such pyrene-labeled HMWSPs.16–18 In order to simplify the synthesis, a novel fluorescent label, vinylcarbazole, might be employed, since the HMWSPs could be labelled simply by copolymerization with such a commercial monomer. Unfortunately those spectroscopic features of the pyrene-labeled system could be hardly generalized to other systems because the fluorescence characteristics of organic molecular aggregates vary from system to system. Quantum yields or spectral details might be influenced differently by aggregation processes due to new excited species formation upon aggregation, such as excimers7 or different kinds of aggregates.8–11 For instance, aggregation usually induced quenching of fluorescence while aggregates of some non-emissive monomers could exhibit fluorescence with high quantum yields due to aggregation-restricted intramolecular rotation,19 which is the so-called aggregation-induced emission phenomenon reported by Tang et al.20,21 Although the interesting fluorescence of poly(N-vinylcarbazole) (PNVC) has attracted considerable attention for a long time,22–28 fluorescence spectral signatures of association of carbazole units were rarely reported. It has been found that two structured emission peaks transformed to a broad structureless band once the monomers were polymerized.23–25 Such a phenomenon has also been observed for several copolymers of N-vinylcarbazole (NVC) systems without detailed investigation of the transition process.26–28 So far all the carbazole-containing copolymers studied were totally hydrophobic and the medium was an organic solvent known as a good solvent for carbazole units, and to the best of our knowledge carbazole-labelled HMWSPs have not been reported. As a result little is known of the fluorescence characteristics of association behaviors of carbazole units in water, which is of significance in fluorescence analysis for such new labelled systems.
For this purpose a novel labelled HMWSP was prepared by copolymerization of acrylamide (AM) and NVC in this work. Carbazole units were introduced into the hydrophilic polyacrylamide main chain, which played roles of both hydrophobic units and fluorescence label. A concentration-dependent association process of this carbazole-labelled polyacrylamide in water has been investigated by light scattering and fluorescence spectroscopy. The spectroscopic features of the excitation and emission spectra reveal three distinct regimes for the association process, which could be employed to analysis the structural transition of carbazole-labelled HMWSPs.
Experimental section
Materials
AM was purchased from Jiangxi Changjiu Biological and Chemical Corp. (China). NVC was purchased from Xiya Chemical Corp. (China). Ceric ammonium nitrate and sodium bisulfite were purchased from Kelong Chemical Corp. (China). All chemicals were used without purification.Synthesis of carbazole-labeled polyacrylamide
The carbazole-labeled polymers were synthesized by dispersion polymerization. Settled amounts (Table 1) of NVC and AM were dissolved in ethanol at first and a redox initiation system composed of ceric ammonium nitrate and sodium bisulfite was adopted to initiate the copolymerization at room temperature (∼25 °C) for 10 h. The products were obtained by filtration of the reaction solution. For further purification, the polymers were washed with ethanol to remove the unreacted monomers and dried in vacuum for 24 h at 50 °C.| Sample | AM/g | NVC/g | Ethanol/g | Mwa/kDa | PDIa | fnvc/% | Fpnvcb/% |
|---|---|---|---|---|---|---|---|
| a The relative molecular weight (Mw) and polydispersity index (PDI) were analyzed by GPC.b The mole fraction of PNVC (Fpnvc) was analyzed by 1H NMR. | |||||||
| PAM-r-PNVC02 | 25 | 0.2 | 55 | 145.9 | 2.32 | 0.45 | 0.28 |
| PAM-r-PNVC03 | 25 | 0.3 | 55 | 145.8 | 2.13 | 0.55 | 0.42 |
| PAM-r-PNVC05 | 25 | 0.5 | 55 | 132.7 | 2.19 | 0.61 | 0.70 |
Characterization
Dynamic light scattering (DLS) experiments were performed with a commercial laser light scattering spectrometer (Brookhaven BI-200SM) equipped with a digital time correlator (BI-9001) and a cylindrical 35 mW He–Ne laser (λ = 532 nm, uniphase) as light source. The decay rate (Γ) of the aggregates was obtained by Contin analysis29 of the experimental correlation function. Combining the equation Γ = Dq2 and Stocks–Einstein equation Rh = kbT/(6πηD), the size of the aggregates as well as its distribution could be obtained (where the scattering vectors q = (4πn/λ) × sin(θ/2), where n, λ, and θ are the solution refractive index, wavelength of the incident light, and the scattering angle, while kb, T, η, D, and Rh are the Boltzmann constant, absolute temperature, viscosity of the solvent, diffusion coefficient, and hydrodynamic radius of the aggregates respectively). The scattering angle was fixed at 90°.
All the excitation and emission spectra were recorded with a fluorescence spectrometer (Horiba-Jobin-Yvon, Fluoromax-4). Band pass used for recording fluorescence and fluorescence excitation spectra was 1 nm.
Results and discussion
Synthesis and characterization of carbazole-labeled polyacrylamide
As a matter of fact, copolymers of NVC and AM have been less reported due to two difficulties of the copolymerization. First, NVC is a typical kind of non-conjugated monomer which was thought to be not easily copolymerized with conjugated monomers such as styrene, AM etc. by radical polymerization.30,31 However, the latest research32 demonstrated that NVC is not so less active as thought and its copolymers with other conjugated monomers might be synthesized. The second difficulty is the mismatch of their solubilities. AM as well as corresponding polymers are water soluble and therefore water was the most adopted reaction medium for polymerization of AM. Contrarily NVC and PNVC couldn't be soluble in water. For this reason ethanol was adopted as reaction medium to dissolve both monomers NVC and AM in our experiments. Because high molecular weight PAM exhibited limited solubility in ethanol, the ultimate copolymers would precipitate from the reaction solution.From the NMR results in Fig. 1a it could be confirmed that the NVC had been copolymerized with AM successfully according to the characteristic peaks of PNVC (aromatic protons of carbazole units at 8.3 to 6.8 ppm in Fig. 1b). Based on the calculation and comparison of peak areas of different characteristic peaks from the two components, we can find that the copolymer exhibits a higher mole fraction of PNVC when the fraction of NVC in the monomer feed was increased. In addition there is a slight effect of the monomer composition on the molecular weight from the GPC results in Table 1. All three samples are of nearly the same molecular weight. A possible reason might be that this molecular weight is the upper limitation for solubility. Although mole fraction of carbazole units is rather low (<1%), obvious associative behaviors could be found when certain amounts of polymer were dissolved in water. Meanwhile efforts have been made to increase the mole fraction of carbazole units, while higher contents of carbazole units would worsen the modified PAM solubility in water due to the extreme hydrophobicity of PNVC. It should be stressed that the NVC was copolymerized rather than blended in the sample, which was supported by the emission peaks of vinylcarbazole monomer instead of the structureless peaks of aggregates (in fluorescence characterization section) and comparison between the emission spectra of the copolymer and that of NVC monomer in water (S1†).
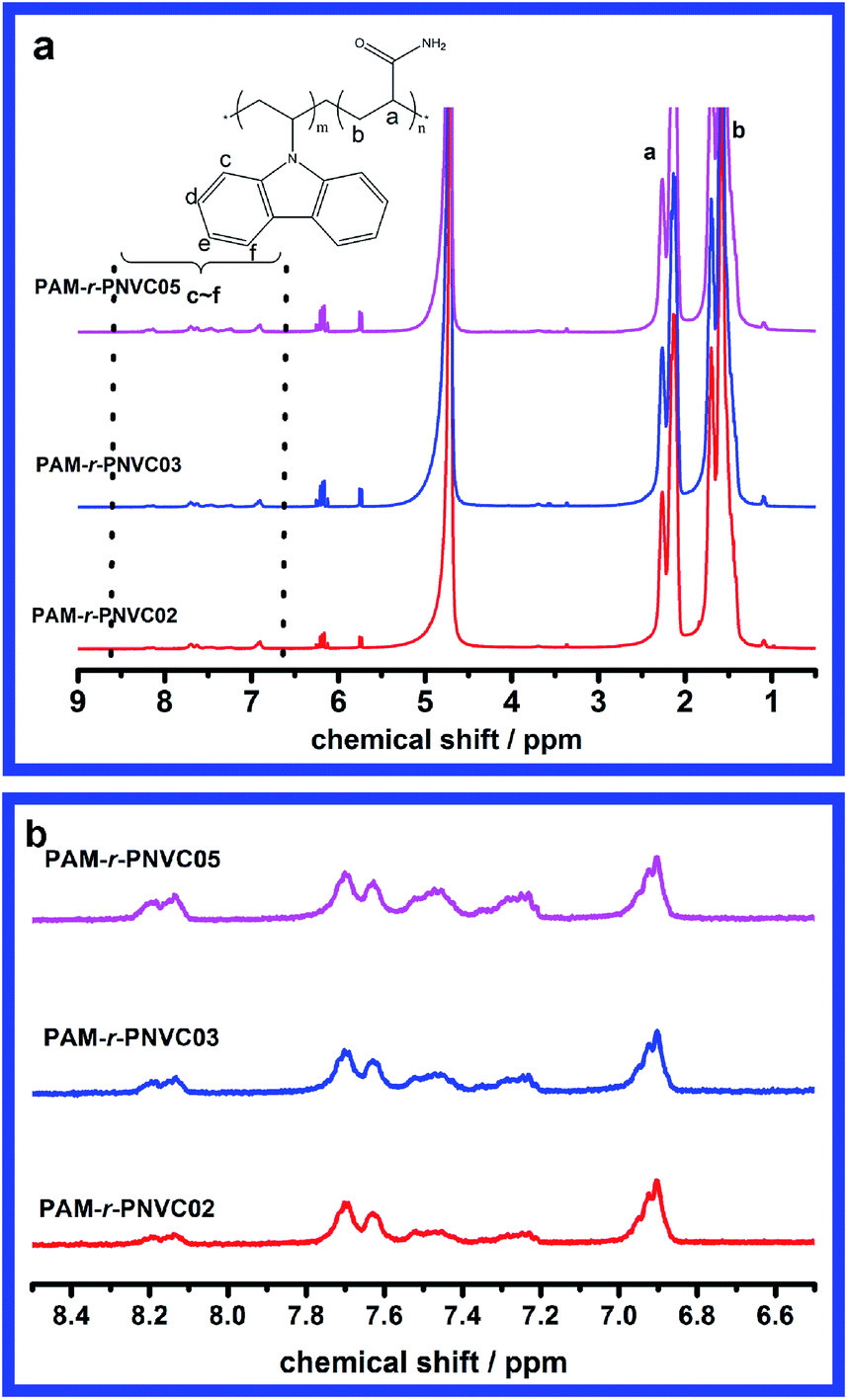 | ||
| Fig. 1 1H NMR spectra of the copolymers of acrylamide and N-vinylcarbazole (a) and a close-up view of the characteristic peaks of carbazole units (b). | ||
Concentration-dependent association process
Concentration-induced association might be the most notable character of amphiphilic polymers, including HMWSPs, which was widely studied by various groups.1,2,5,6,17 For a HMWSP, when the concentration was beyond the critical micellization concentration, unimers of the HMWSP would associate to form micelles. Further increase in concentration would induce formation of links between different micelles which might result in a distinct rise in viscosity.33 This concentration-induced association also held true for our carbazole-labeled PAM. From DLS results in Fig. 2, it could be found that this kind of HMWSP (PAM-r-PNVC05) formed aggregates in water when the concentration was above 1000 ppm. At lower concentration it was difficult to obtain a reliable correlation function because of the rather low scattering intensity. Thus we can determine the critical concentration for PAM-r-PNVC05 is near 1000 ppm. According to the size distribution in Fig. 2, it could also be concluded that larger aggregates were formed at higher concentration. The average diameter is about 28.1 nm at a concentration of 1000 ppm while the value increases to 86.7 nm, 172.9 nm and 251.9 nm when the concentration increases to 2000 ppm, 3000 ppm and 5000 ppm respectively. Therefore, different from that in block copolymers and some other associative polymers, an open association model might be more suitable to describe the aggregation behaviours of PAM-r-PNVC05, which means the association level, or in other words the aggregation number, is determined by the concentration. | ||
| Fig. 2 Hydrodynamic diameters of PAM-r-PNVC05 as well as their distribution at different concentrations obtained from DLS. | ||
Fluorescence study of the aggregation process
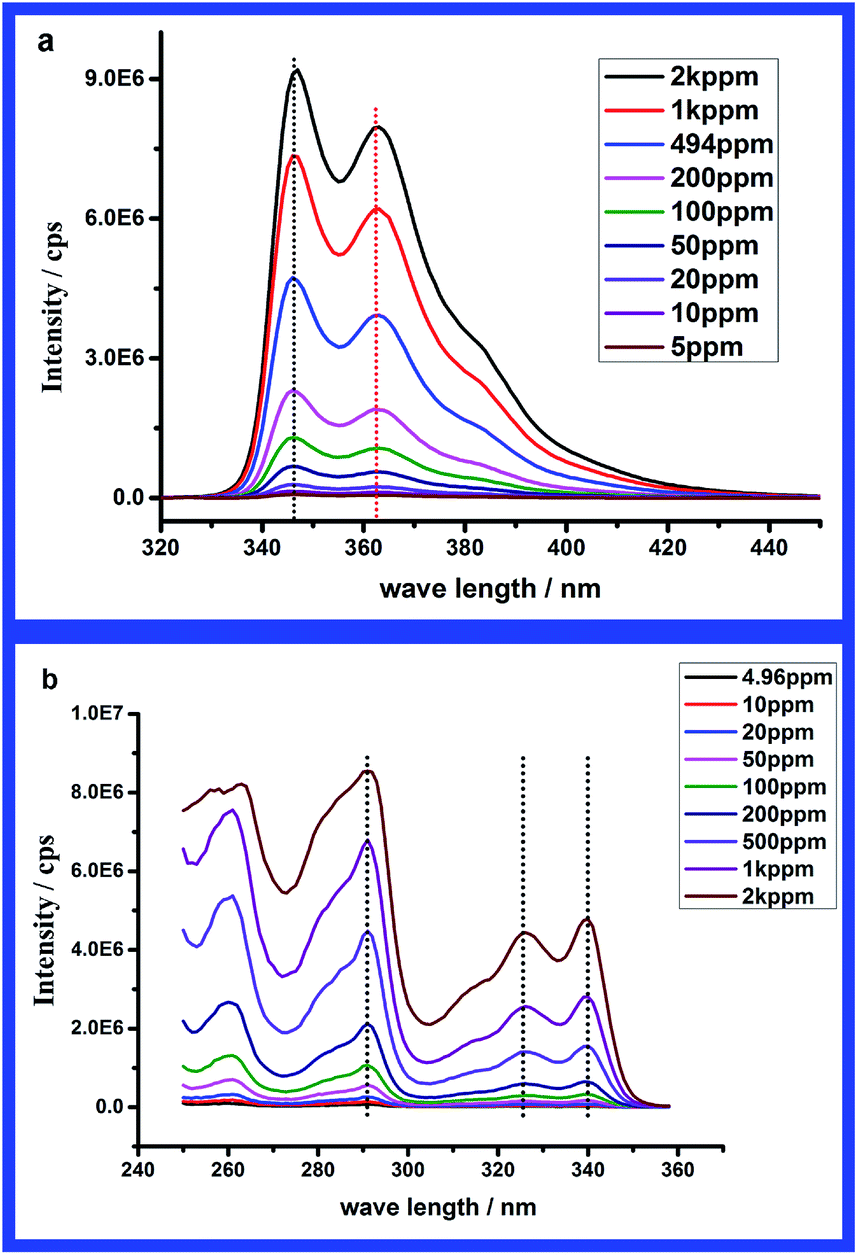
As the concentration-dependent aggregation behaviour was well established in this system, a fluorescence study of the association process of PAM-r-PNVC05 was executed to reveal the fluorescence spectral features of carbazole units upon aggregation. Fig. 3a gives the overall emission spectra of PAM-r-PNVC05 for a large range of concentration from 5 ppm to 200 kppm. The excitation wavelength was fixed at λexcitation = 291 nm, because the highest intensity could be acquired at this wavelength according to either the literature24,27,28 or our experiment in dilute solution. However, it seems too complicated to summarize the variation of the spectra in one or two words, because it is sensitively dependent on the concentration. For different concentration regimes, different laws govern the change of emission spectra. For simplicity the intensity of the second emission peak at 363 nm (0–1 transition) in the spectra is plotted versus concentration in Fig. 3b. According to the variation tendency in Fig. 3b, the overall concentration range was divided roughly into three different regimes. In regime (I), the emission intensity increases monotonically to a maximum, while it decreases rapidly to a minimum in regime (II), followed by a slight increase in regime (III). | ||
| Fig. 3 (a) 3D plot and projection of emission spectra and (b) the intensity of the second emission peak (λ = 363 nm) of PAM-r-PNVC05 in water at different concentrations (λexcitation = 291 nm). | ||
 | ||
| Fig. 4 Emission spectra (λexcitation = 291 nm) (a) and excitation spectra (λemission = 363 nm) (b) of PAM-r-PNVC05 in water at different concentrations in regime (I). | ||
In order to analyse the intensity variation, the intensities of corresponding characteristic peaks in emission spectra and excitation spectra were plotted versus concentration. In both Fig. 5a and b, a linear relationship between the intensity and concentration could be found at low concentration. Because both emission and excitation are only related to the number of chromophores, we speculate that only unimers exist in this regime, which also coincides with the results from DLS. From Fig. 5a, the intensity deviates from the linear relationship when the concentration is beyond 500 ppm. Similar deviation from linearity is also found for the characteristic peak at λ = 291 nm in Fig. 5b, while the linear relationship covers a wider range for the lower energy peaks. According to the analysis, regime (I) is assigned to unimer regime at concentrations lower than 1000 ppm.
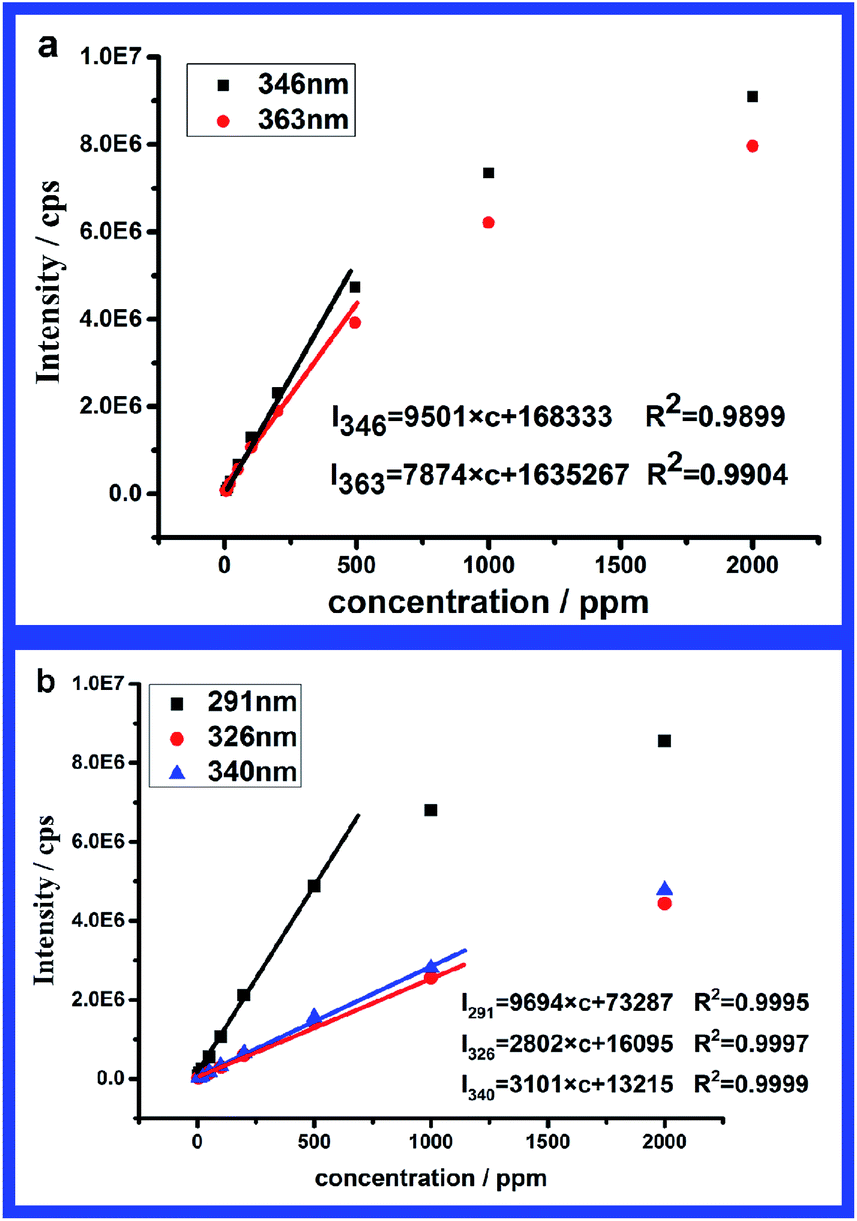 | ||
| Fig. 5 Intensity of characteristic peaks in emission spectra (a) and excitation spectra (b) at various concentrations in regime (I). | ||
 | ||
| Fig. 6 Emission spectra (λexcitation = 291 nm) (a) and excitation spectra (λemission = 363 nm) (b) of PAM-r-PNVC05 in water at different concentrations in regime (II). | ||
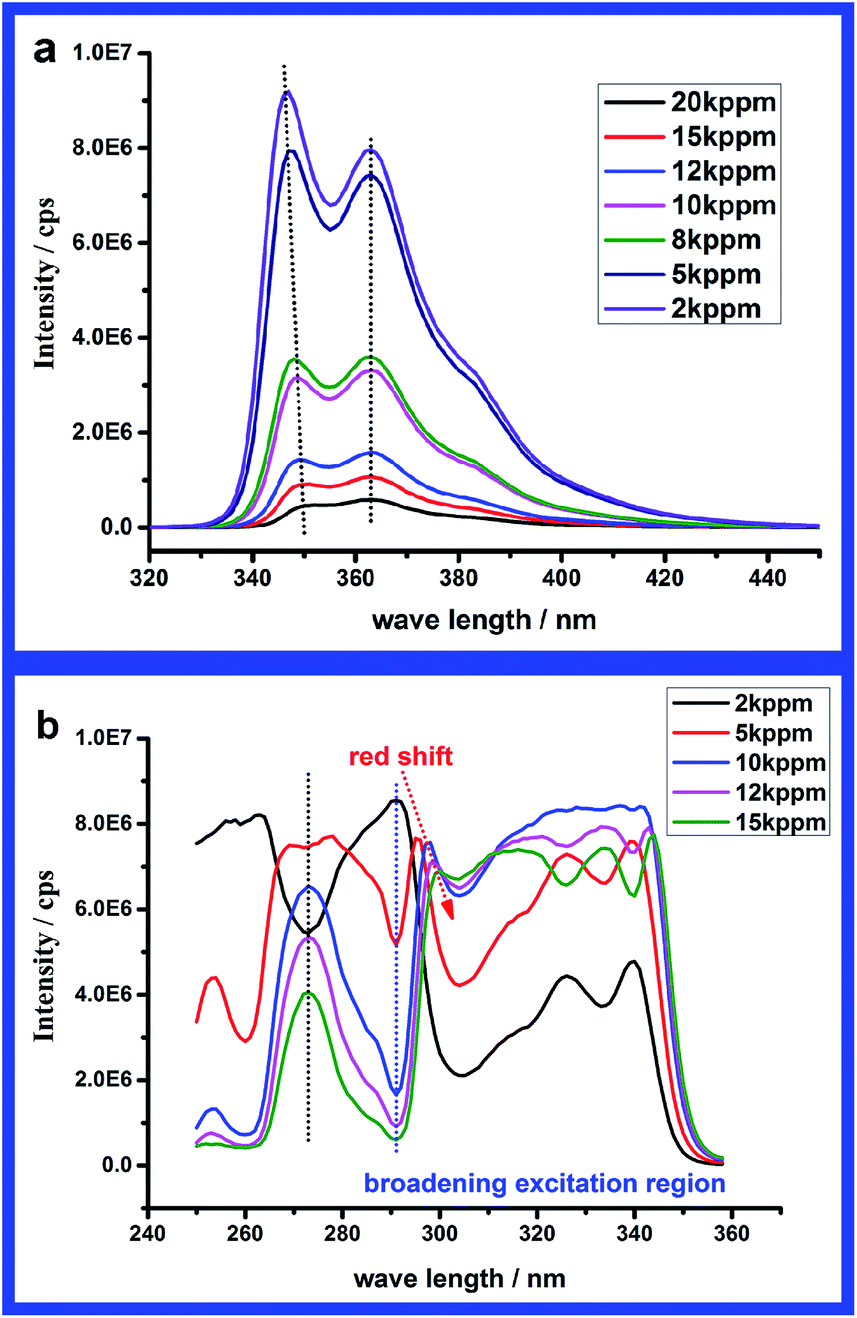
Besides the fluorescence intensity, both emission and excitation spectral shapes vary with change in concentration in this regime. For the emission spectra, there are two distinct changes. First is a slight red shift of the higher energy emission peak, from 346 nm at 2 kppm to 352 nm at 20 kppm. However, the lower energy emission peak remains at 363 nm for the whole concentration range. Another obvious change in emission spectra is that the relative intensity of the higher energy emission peak decreases with increasing concentration. From Fig. 7a we can find that the intensity ratio of the two emission peaks I346/I363 (the wavelength of higher energy emission peak is slightly changed according to Fig. 6a) keeps nearly constant at concentrations below 1000 ppm and decreases from 1.2 to 0.8 when the concentration increases to regime (II). This ratio might be an effective parameter to estimate the aggregation of carbazole units. As to the excitation spectra, the relative intensity of the lower energy band (300–350 nm) increases with increasing concentration. The intensity ratios of characteristic peaks of the excitation spectra have also been plotted versus concentration in Fig. 7b. The intensity ratios (I326/I291 and I340/I291) between the higher energy peak (270–300 nm) and lower energy peaks (300–350 nm) keep constant in the unimer regime (c < 1000 ppm) and increase when the carbazole units begin to aggregate. On the contrary, the intensity ratio (I326/I340) of the two characteristic peaks in the lower energy band changes little. When the concentration is above 2 kppm, an obvious red shift of the higher energy absorption band could be found in Fig. 6a. Due to the red shift of the higher energy band and the intensity increase of the lower energy band, the two bands merge to form a broad excitation region (290–350 nm), which results in a change in wavelengths corresponding to the peak and valley. In the broad excitation region, the distinction between peak and valley becomes rather indistinct especially when the concentration is above 10 kppm. Comparing with the absorption spectra in Fig. 8a, we can find the absorption spectrum at lower concentration (c < 2000 ppm) is nearly identical in shape to the excitation spectrum in Fig. 6b. While the absorption spectra and excitation spectra are no longer superimposable at higher concentration (c > 2000 ppm), which usually implies that several species were present or a sole species existed in several different forms in the ground state, such as aggregates.34 Considering the DLS results, higher concentration induced larger particles, because aggregation of the fluorescent carbazole units influences the energy level, with a large conjugated system making the electrons more localized. For this reason all these spectral changes are mainly ascribed to the slight aggregation of carbazole units. Further analyzing the relative absorption intensity of different bands in Fig. 8b, we can find the proportion of S0 → S2 absorption (270–300 nm) decreases and that of S0 → S1 absorption (300–350 nm) increases with aggregation. The relative ratio (I292/I340) of S0 → S2 absorption to S0 → S1 absorption decreases from 3.2 to 1 when concentration increased from 1000 ppm to 10 kppm. This implies that the change in Fig. 7b resulted from aggregation-induced change in absorption bands. Additionally it should be noted that S0 → S2 transition still absorbed more than S0 → S1 transition but emitted much less upon aggregation. This might be ascribed to quenching of new formed aggregates to the unimer. However, we still could not exclude the influence of the inner filter effect completely.
 | ||
| Fig. 7 Fluorescence intensity ratios of different characteristic peaks in emission spectra (a) and excitation spectra (b) at different concentrations. | ||
 | ||
| Fig. 8 Absorption spectral (a) and absorption intensity ratio of different characteristic peaks at different concentrations (b) in regime (II). | ||
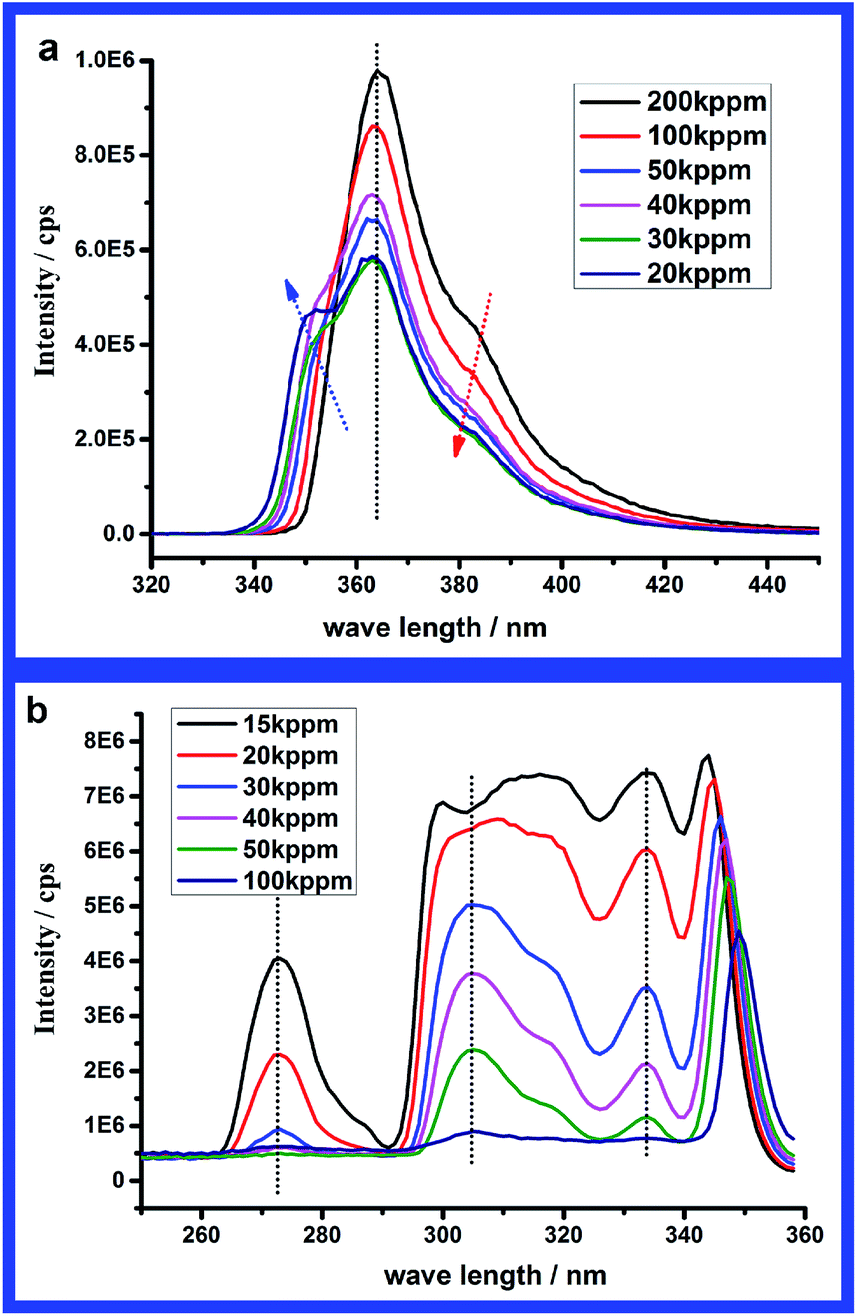 | ||
| Fig. 9 Emission spectra (λexcitation = 291 nm) (a) and excitation spectra (λemission = 363 nm) (b) of PAM-r-PNVC05 in water at different concentrations in regime (III). | ||
Association-induced fluorescence features of carbazole units
Despite the description of the spectral signatures of carbazole aggregates in the previous section, an attempt will be made for the fluorescence features to be interpreted and understood within the scope of organic molecular aggregates.35 Generally speaking, once organic molecules become close enough to each other, both absorption and fluorescence spectra might deviate from those of monomers because of the dipole–dipole coupling between the transition dipole moments of the neighboring molecules.36 According to different orientations of the chromophores within the aggregates, the aggregates could be classified as H-aggregates and J-aggregates ideally. When the aromatic molecules pack in a side-by-side orientation to form H-aggregates, the excitonic coupling is positive leading to a blue-shift absorption and quenched fluorescence.8,10,11,37,38 While for J-aggregates the molecules pack in a head-to-tail orientation and the coupling is negative resulting in a red-shift absorption.9,38–40 Thus from the excitation spectra of PAM-r-PNVC05 it seems that J-aggregates were formed by carbazole units, because an obvious red-shift exists in the excitation spectra of the aggregates. Additionally in Spano's work36 another important spectral feature of J-aggregates was that the ratio of the oscillator strengths in the A1 to A2 bands would increase upon J-aggregation, where A1 and A2 donated a proportion of S0 → S1 transition and S0 → S2 transition. This feature could be found in Fig. 8b, as the peaks at 327 nm and 340 nm in the absorption spectra represent S0 → S1 transition while the peak at 292 nm represents S0 → S2 transition. As stated by Spano,36 in the strong coupling regime oscillator strength was mainly concentrated in a single lower energy peak, which coincided with the results in Fig. 9b. Thus all the excitation and absorption spectral features suggest that J-aggregates were formed by carbazole units.Unfortunately the observed aggregation-induced quenching in the carbazole aggregates is contrary to that of J-aggregates, which is more likely ascribed to H-aggregates. Meanwhile, the 0–0 transition would vanish upon H-aggregation which is opposite to J-aggregation. From the decrease in I346/I363 (peak at 346 nm represents the 0–0 transition and peak at 363 nm represents the 0–1 transition) in Fig. 7a and vanishing of emission peak at 346 nm in Fig. 9a, the spectral signatures of emission imply the vanishing of the 0–0 transition upon aggregation. Thus it seems H-aggregates were formed by carbazole units according to the emission spectra. However, the same vanishing of the 0–0 emission might also be observed in J-aggregates with increasing disorder according to Spano's work.36 Thus the spectral signatures of excitation and emission could be well understood within the scope of Spano's simulation results,36 and a possible explanation might be that J-aggregates were formed by carbazole units and the disorder in the aggregates increased upon the open association process from unimers to slight aggregation in regime (II) to strong aggregation in regime (III).
The speculation of disorder in the J-aggregates could also be supported by the absence of excimer formation from the emission spectra. An excimer41,42 denotes molecular configurations which absorb as monomers but in the excited state one excited molecule and one un-excited monomer physically associate to form dimers and fluoresce as such. It was very common to observe excimer formation in aromatic chromophores7,42 such as pyrene and carbazole at high local concentration. The excimers formed by carbazole-containing polymers have been also investigated in dilute solution and thin film for a long time.12,23–28,43 Different excimer species have been observed which were assigned as partially overlapped (lower energy λemission = 380 nm) and sandwich excimers (higher energy λemission = 430 nm). It is reasonable that there is only monomer emission in regime (I) because of the extremely low mole fraction of NVC in the copolymer which is the same as the situation in the literature. Unexpectedly, there are no obvious signatures of excimer emission upon aggregation in regimes (II) and (III). The most probable reason might be the disorder of carbazole units in the aggregates. Only for the condition that the molecules in the crystal must be ordered pairwise in parallel planes, with a small distance between neighboring planes, might an excimer form in the aggregates.35 However, for carbazole units in the aggregates, they lose their mobility to adjust to the right conformation for excimer formation. Therefore the dominating head-to-tail orientation and the disorder in the aggregates caused the absence of excimer formation in our system.
Conclusions
In summary, a novel carbazole-labeled HMWSP was synthesized by dispersion copolymerization of NVC and AM in ethanol and a concentration-dependent open association process of this labeled polymer in water has been studied by DLS and fluorescence spectroscopy. The spectral features of excitation and emission spectra were summarized in three different regimes for association process. All these spectral signatures are tried to be understood within the scope of Spano's simulation and carbazole units are ascribed to formation of J-aggregates with high disorder. However, these explanations need to be further investigated since the inner filter effect could hardly be excluded experimentally, especially as regards the signatures in excitation spectra. Nevertheless, these fluorescence features in the association process of carbazole units were of significance to develop a carbazole-based fluorescence analytical method and fluorescent materials.Acknowledgements
We appreciate the financial support of this research by the National Natural Science Foundation of China (major national science and technology projects: 2011ZX05011-004).Notes and references
- L. Karlson, Hydrophobically Modified Polymers. Rheology and Molecular Associations, Lund University, 2002 Search PubMed.
- J. Sprakel, Physics of Associative Polymers: Bridging Time and Length Scales, Universiteit, 2009 Search PubMed.
- D. Wever, F. Picchioni and A. Broekhuis, Prog. Polym. Sci., 2011, 36, 1558–1628 CrossRef CAS.
- K. C. Taylor and H. A. Nasr-El-Din, J. Pet. Sci. Eng., 1998, 19, 265–280 CrossRef CAS.
- M. Rubinstein and A. V. Dobrynin, Trends Polym. Sci., 1997, 5, 181–186 CAS.
- M. A. Winnik and A. Yekta, Curr. Opin. Colloid Interface Sci., 1997, 2, 424–436 CrossRef CAS.
- S. A. Jenekhe and J. A. Osaheni, Science, 1994, 265, 765–768 CAS.
- F. Panzer, M. Sommer, H. Bässler, M. Thelakkat and A. Köhler, Macromolecules, 2015, 48, 1543–1553 CrossRef CAS.
- T. E. Kaiser, V. Stepanenko and F. Würthner, J. Am. Chem. Soc., 2009, 131, 6719–6732 CrossRef CAS PubMed.
- H. V. Berlepsch and C. Böttcher, J. Phys. Chem. B, 2015, 119, 11900–11909 CrossRef PubMed.
- N. Nizomov, E. N. Kurtaliev and S. I. Rahimov, J. Mol. Struct., 2012, 1029, 142–148 CrossRef CAS.
- J. Grazulevicius, P. Strohriegl, J. Pielichowski and K. Pielichowski, Prog. Polym. Sci., 2003, 28, 1297–1353 CrossRef CAS.
- S. Maruyama, H. Suzuki, X.-t. Tao, T. Wada, H. Sasabe, S. Miyata and T. Kamata, Phys. Chem. Chem. Phys., 2000, 2, 3565–3569 RSC.
- J. Garcia-Amorós, S. Swaminathan, Y. Zhang, S. Nonell and F. M. Raymo, Phys. Chem. Chem. Phys., 2015, 17, 11140–11143 RSC.
- W. Klöpffer, J. Chem. Phys., 1969, 50, 2337–2343 CrossRef.
- S. A. Ezzell, C. E. Hoyle, D. Creed and C. L. McCormick, Macromolecules, 1992, 25, 1887–1895 CrossRef CAS.
- M. C. Kramer, C. G. Welch, J. R. Steger and C. L. McCormick, Macromolecules, 1995, 28, 5248–5254 CrossRef CAS.
- P. Deo, N. Deo, P. Somasundaran, S. Jockusch and N. J. Turro, J. Phys. Chem. B, 2005, 109, 20714–20718 CrossRef CAS PubMed.
- J. Chen, C. C. Law, J. W. Lam, Y. Dong, S. M. Lo, I. D. Williams, D. Zhu and B. Z. Tang, Chem. Mater., 2003, 15, 1535–1546 CrossRef CAS.
- J. Luo, Z. Xie, J. W. Lam, L. Cheng, H. Chen, C. Qiu, H. S. Kwok, X. Zhan, Y. Liu and D. Zhu, Chem. Commun., 2001, 1740–1741 RSC.
- Y. Hong, J. W. Lam and B. Z. Tang, Chem. Commun., 2009, 4332–4353 RSC.
- D. Trivedi and H. Nalwa, Handbook of Organic Conductive Molecules and Polymers, 1997, p. 2 Search PubMed.
- F. De Schryver, J. Vandendriessche, S. Toppet, K. Demeyer and N. Boens, Macromolecules, 1982, 15, 406–408 CrossRef CAS.
- D. Ghosh and N. Chattopadhyay, J. Lumin., 2011, 131, 2207–2211 CrossRef CAS.
- G. Johnson, J. Chem. Phys., 1975, 62, 4697–4709 CrossRef CAS.
- N. Fernández-Peña, T. Carmona, M. P. Tarazona, E. Saiz and F. Mendicuti, Polym. Int., 2011, 60, 1487–1496 CrossRef.
- K. Davidson, I. Soutar, L. Swanson and J. Yin, J. Polym. Sci., Part B: Polym. Phys., 1997, 35, 963–978 CrossRef CAS.
- A. Brar, M. Kaur, M. Balamurli and S. Dogra, J. Appl. Polym. Sci., 2006, 100, 372–380 CrossRef CAS.
- S. W. Provencher, Comput. Phys. Commun., 1982, 27, 229–242 CrossRef.
- G. Moad, E. Rizzardo and S. H. Thang, Aust. J. Chem., 2012, 65, 985–1076 CrossRef CAS.
- M. Benaglia, J. Chiefari, Y. K. Chong, G. Moad, E. Rizzardo and S. H. Thang, J. Am. Chem. Soc., 2009, 131, 6914–6915 CrossRef CAS PubMed.
- D. J. Keddie, C. Guerrero-Sanchez and G. Moad, Polym. Chem., 2013, 4, 3591–360125 RSC.
- K. C. Taylor, Annu. Trans. - Nord. Rheol. Soc., 2003, 11, 13–20 CAS.
- B. Valeur and M. N. Berberan-Santos, Molecular Fluorescence: Principles and Applications, John Wiley & Sons, 2012 Search PubMed.
- M. Schwoerer and H. C. Wolf, Organic Molecular Solids, John Wiley & Sons, 2008 Search PubMed.
- F. C. Spano, Acc. Chem. Res., 2009, 43, 429–439 CrossRef PubMed.
- S. Basak, N. Nandi, K. Bhattacharyya, A. Datta and A. Banerjee, Phys. Chem. Chem. Phys., 2015, 17, 30398–30403 RSC.
- H. V. Berlepsch, K. Ludwig and C. Böttcher, Phys. Chem. Chem. Phys., 2014, 16, 10659–10668 RSC.
- I. F. Pierola and I. E. Pacios, J. Fluoresc., 2012, 22, 145–150 CrossRef CAS PubMed.
- R. Teixeira, S. M. Andrade, V. Vaz Serra, P. M. Paulo, A. Saánchez-Coronilla, M. G. Neves, J. A. Cavaleiro and S. M. Costa, J. Phys. Chem. B, 2012, 116, 2396–2404 CrossRef CAS PubMed.
- T. Förster, Angew. Chem., Int. Ed. Engl., 1969, 8, 333–343 CrossRef.
- J. Birks, Rep. Prog. Phys., 1975, 38, 903 CrossRef CAS.
- C. David, M. Piens and G. Geuskens, Eur. Polym. J., 1972, 8, 1291–1297 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c6ra15898f |
| This journal is © The Royal Society of Chemistry 2016 |